What is design control for medical devices and how does it work?
What is design control? It's a common question for medical device quality professionals just starting out on their journey to market. Navigating the medical device regulatory environment can seem daunting at times, and it's easy to get lost in technical jargon and complex standards. Taking a step back to understand how a requirement can be beneficial to your bottom line is a helpful approach to managing regulatory overwhelm.
For those that are new to the regulatory landscape, design controls is one of the more complex set of procedures to comprehend. Design control guidance for medical device manufacturers is therefore an important piece of information to brush up on as you plan and prepare your medical device quality management system.
This is your complete guide to understanding just what design controls really means for your medical device product development.
Table of Contents
What are design controls?
Design controls for medical devices are, in short, a series of structured requirements that facilitate a compliant design and development process.
They act as 'checkpoints', in place to ensure your device is safe and effective when brought to market. If you are a medical device manufacturer, compliance with design controls is required - and FDA design control guidance in FDA 21 CFR 820.30 is a good place to start as you look to understand what design controls are.
In the world of medical devices and SaMD solutions, 21 CFR 820.30 design controls help FDA-regulated organizations accomplish five critical objectives:
- Deliver quality products
- Ensure user safety
- Maintain regulatory compliance
- Keep costs down
- Accelerate time to market
What is the purpose of design controls?
Design controls are required to improve product quality and help companies avoid manufacturing defects that can lead to recalls or other significant issues.
Complying with the requirements fully can prevent recalls and other regulatory enforcement actions. FDA warning letters and recalls are outcomes medical device or software as a medical device (SaMD) companies want to avoid, underscoring the need for a strong design controls system.
Design controls provide a structure for ensuring that device design and development is completed in a highly controlled manner. This means that each design step is planned before being executed and that the execution is verified.
Design controls also ensure that a cross-functional approach is taken to the design process so that additional viewpoints are considered and potential roadblocks are realized early.
Ultimately, organizations use these controls to ensure their design processes deliver products to market that are safe and effective.
RELATED READING: Ultimate guide to medical device design controls
How do design controls factor into a quality management system?
Design controls is a critical process that should be integrated into your quality management system (QMS). Your QMS should also have procedures in place to manage documents, records, and suppliers, so design controls link those pieces together with additional development specific processes.
The design and development process, which design controls regulate, extends beyond creating and testing a device.
Creating design control documentation, ensuring that suppliers are adequately qualified for their role in the manufacturing process, and maintaining records are just a few of the QMS procedures that directly feed into design controls. Once the initial design has been completed and brought to market, the QMS will continue to be necessary for ongoing compliance. Regulators require ongoing monitoring and quality system reporting related to the device including complaint handling, nonconformance monitoring, and Risk Management.
Even when the design controls process has been completed, the work is far from done and you will need to rely on your existing QMS for ongoing control of your device.
Design control guidance for medical device manufacturers
Now that we understand how design controls fits into the big picture, we can dive into the process and the individual design control phases themselves.
The following elements are a generalized approach of blended FDA and ISO requirements, mostly using FDA terminology.
Each element of design controls is carefully considered by an auditor, and those specific considerations are listed at the conclusion of each of the following design controls elements.
1. Establish QMS procedures
Starting with effective, well-thought out QMS procedures will make for a smoother implementation.
Here's a medical device design control process flow chart to help you visualize how design controls should slot into your wider medical device quality management system.
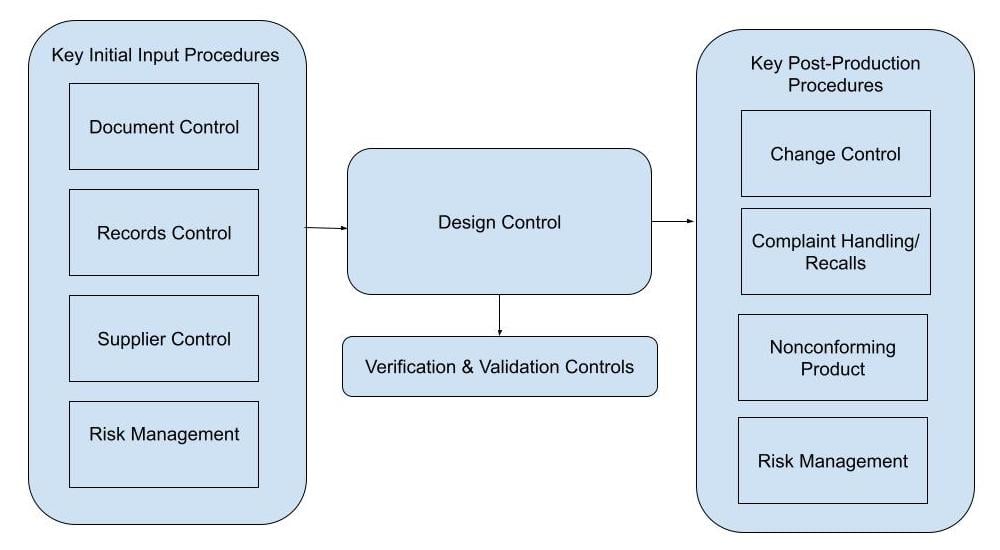
When an organization develops a device before establishing the QMS and design controls process, some common issues that may arise are:
- Missed compliance steps in device development
- Ineffective procedures (written out of context)
- Disorganization and more resources used in retrospective "clean-up"
- Messy audits (for all the above reasons)
- Delays getting product to market
- Design changes to device that does not meet all requirements
Writing procedures is only half of the work—the procedures must also be implemented. You can have volumes of procedures to show to an auditor, but if you can’t show that they’ve been implemented you're not in compliance.
Auditors look for: Are the activities performed by the organization regarding design controls consistent with the procedures developed (i.e. are they implemented)?
2. Document design and development planning
Once you have your QMS and design control procedures in place, you need to develop your design plan and your design control documentation. Typically organizations create development plans with 'phases' or 'stages' that align roughly with these milestones, which we'll explore in more detail. Those phases or stages are:
- User Needs / Design Input / Initial Risk Assessment
- Design Output / Device Risk Assessment
- Design Manufacturing Process / Supplier Qualification / Process Risk
- Design Verification and Design Validation / Design & Process Risk
- Design Transfer for Manufacturing
Auditor looks for: Are planning documents established? Are team members defined? Are plans effective and being followed?
3. Apply risk management
Risk management is a critical part of all areas of design and development, and will be summarized here.
Risk management is most commonly applied per the ISO 14971 standard, recognized by both the FDA and EU. It is applied early to the development process, as risks and harms are to be considered as an outcome and a driver for User Needs and Design Inputs. Risk is subsequently re-evaluated as the design takes shape and new considerations likely arise. Risk will be continually reviewed throughout the product life cycle.
Auditor looks for: Is risk management applied to the design and development process throughout? Are risks mitigated through the design process?
4. Perform design reviews
Design reviews are checkpoints throughout the design process that can prevent wasted time and money on a device that won’t work or meet requirements. Completing thorough, cross-functional reviews at each phase and taking necessary corrective actions can prevent issues down the road when it comes time for design verification and validation (VnV).
Regulations don't mandate the specific project milestones at which design review is to occur, so this is up to the organization to define in their SOPs. The following is an example of what's common in industry:
- Phase 1 Close-out: User Needs/Design Input Review
- Phase 2 Close-out: Design Output Review
- Phase 3 Close-out: Design Verification and Validation Results Review
- Phase 4 Close-out: Design Transfer/Release Final Review
Primary expectation in Design review:
- Record attendees of the review, their roles, and approval of the review
- Record the stage/phase being reviewed
- Record meeting minutes with any action items
- Ability to connect the review to the respective device's Design History File (DHF)
Auditors look for: Does each design review include an independent reviewer not directly involved with the design and development project? Are action items being addressed that are noted during phase reviews? Are phases being used to gate the design from proceeding?
5. Establish user needs and design inputs
Design input requirements are an important area of your design control activity. Before device development begins, the purpose of the device must be defined to ensure it achieves that intended purpose for patients and medical operators.
User needs and design inputs are very similar. Notice the distinction in the example for a syringe device:
- User Need example: The syringe must fit in an emergency medical services go-bag
- Typically driven by use case marketing feedback from potential users
- Finished device will be Validated later in the process to ensure the user is satisfied with the size of the syringe
- Design Input example: The syringe shall be no more than 5" in length and 1" in diameter
- Typically translated from User Needs to something more tangible for the engineering team to work with
- Finished Device will be Verified later in the process to ensure the syringe length/diameter meets the Design Input
Auditor looks for: Are User needs and inputs concise and unambiguous? Do they conflict with one another?
6. Establish design outputs
Design outputs represent the characteristics of the device that make the design inputs and user needs a reality.
Continuing with the example of the syringe device:
- User Need example: The syringe must fit in an emergency medical services go-bag
- Design Input example: The syringe shall be no more than 5" in length and 1" in diameter
- Design Output example: McKesson Brand #16-S1C, 1cc, 4.1" x 0.375" dia. syringe (device drawing)
- The Design Output can be viewed as the answer to the Design Input
- Verification ensures the Design Output meets the Design Input
Auditors look for: Are outputs linked to respective inputs and verification activities in the Design History File? Are the linked drawings and specification files current and controlled documents?
7. Perform design verification and validation (VnV)
VnV are testing activities required for ensuring the device meets the pre-defined specifications and intended purpose.
Verification vs. validation are commonly mixed up:
- Design Verification: Did you design the device right?
- Evaluating if Design Inputs are achieved - are specifications met?
- Design Validation: Did you design the right device?
- Evaluating if User Needs are met
The critical factors of verification and validation are:
- Planning: VnV is complex, and requires qualified individuals performing well-defined tests.
- Samples: VnV must be performed on finished device "equivalents" and the organization must provide rationale for the sample size tested.
- Many organizations cut corners here and end up having to perform multiple iterations of VnV
- Sample sizes must be supported with a rationale statement based on statistical sampling plans
- Software device sample size of 1 is commonly acceptable with proper rationale
Making changes to the device after VnV is costly since it may require going back and completing VnV activities again. Making sure prior phase reviews have been completed can help prevent sending a device out for VnV that is destined to fail.
Auditors look for: Are VnV performed on finished device equivalents? Are sample sizes used based on statistical techniques? Have all user needs and inputs been tested? If you have a contract manufacturer, do you have proof they have validated their manufacturing process? Are all VnV activities documented in the DHF?
8. Document design transfer
Design transfer is the process of making all the final arrangements for transferring the device to commercial production. Once design transfer is completed you should be able to “turn the key” to start manufacturing your product.
Through design transfer you will be able to:
- Connect design documentation to manufacturing documentation
- Verify the Design History File (DHF) is complete and contains all required items
- Document that records of the transfer to manufacturing are maintained
- Ensure that training is completed for manufacturing personnel
- Ensure that regulatory approvals are received before releasing product to the market
Auditors look for: Have all final actions required to send the device to market been taken?
9. Completing design changes and the Design History File (DHF)
The FDA's rule on DHF states: "Each manufacturer shall establish and maintain a DHF for each type of device. The DHF shall contain or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plan and the requirements of this part."
End-users employ various methods of documenting a DHF. Since the regulations include "reference" to the records that make up the file, users will often create a DHF Index that acts as a Table of Contents for DHF records.
So, what is a DHF vs. a DHF Index? They're two varying methods representing the same end-goal. Using a DHF Index to guide the reader through the records that make up the DHF is, without question, a far more effective way to present this complex compilation of records.
DHF:
- Compilation of all design activities related to the development of the device
- Commonly recorded as an index/list of activities with easy access to reference design control documentation listed
- A Design Control Matrix contains a majority of the records found in the Design History File
Design Changes:
- May occur at any time pre/post-release and must be done in accordance with established procedure(s)
- Must follow formal development lifecycle as already prescribed by FDA/ISO (i.e. input, output, VnV, DHF, etc.)
- Require proof of risk assessments related to change - update to risk management file if necessary
Note that Device Master Records (DMR) are commonly mixed up with DHF. The DMR is a "recipe" for the device including drawings, specifications, and work instructions, whereas the DHF represents the activities supporting its development. To make matters more confusing there is also the Device History Record (DHR) which is maintained to house all of the records specific to each batch/lot of devices.
Auditors look for: Does the DHF contain all design activities, or a link to all design activities performed related to a project? Activities include: design reviews, inputs, outputs, verification and validation reports, risk reviews, etc.
Design control checklist
Need a design control checklist to get you on your way?
Download our 6 principles for medical device design control success now.
6 tips for succeeding with design controls
Did you know that design issues are the largest source of medical device recalls in the United States?
Poorly designed devices pose a serious threat to patient health and safety — as well as the long-term success of your growing life start-up or scale-up.
Here are the design control areas that medical device companies most often struggle with - and the things you should pay particular attention to.
1. Don't wait to start
The first question many companies have is: when do design controls begin? Regulation doesn’t explicitly name when design controls are required to start. However, FDA design control guidance provides the following direction:
- Where research ends and design begins
- After feasibility/'proof of concept'
- When you plan to bring your device to market
- Prior to start of any Investigation Device Exemption (21 CFR 812)
- Premarket mechanism of change/revision
Some may argue that starting design controls early will subject their product to greater regulatory burden and cost, but that’s not the case. The reality is that for many companies, starting design controls sooner rather than later can actually save money and time down the road.
Design controls are not vastly more burdensome than following sound engineering practices. A process with well-defined stakeholders, responsibilities and goals is a far better experience for teams than a disjointed approach, regardless of the stage of the product.
Understanding design controls means embracing the discipline needed to successfully develop a compliant medical device or SaMD solution. Beginning design controls early should be driven by the company’s goals to develop a safe and successful product rather than regulatory pressure. The mindset you exhibit toward implementing design controls is just as important as following the regulatory guidance. Building a culture of quality into your company from the outset of the design process will help ensure that quality mindset is maintained throughout the device lifecycle.
2. Go paperless
If you haven’t already done so, migrate from a paper-based system to cloud-based design controls software. These are now the industry standard, as they equip medical device companies to more easily comply with ISO and FDA regulations. A paper-based design control documentation system is cumbersome, manual, and makes it difficult to quickly log information in your DHF or communicate changes across teams.
In a cloud-based system, changes are automatically managed, you and your colleagues can collaborate on design control documentation, design elements can be linked to other areas of your QMS, and more.
Design control software can also assist in the construction of critical elements like your design control traceability matrix, offering a visual digital insight into how your design elements connect with each other.
3. Align quality and product
Often, it can feel like quality and product development are on opposing sides—creating a sense of friction between the two teams. Adopt a unified approach so that quality isn’t seen as a blocker but as a critical function that bolsters the product development team’s goals. Make sure the teams work together and not in silos using different software and project tracking tools.
To support this objective, look for design control software that integrates with typical design tools like Jira, TestRail and AzureAD to demolish silos and friction between the two departments.
4. Plan effectively
Proper planning is a major prerequisite for any effective design control system.
The design planning process begins with a review of design control goals and objectives. For the best results, start by establishing all design control procedures, practices, and systems as far in advance as possible.
Ask yourself these questions:
What activities will we follow to produce highly effective, safe, and compliant medical devices?
How will we manage and enforce these unique design controls?
The second phase of the design control planning process requires quality teams to properly document all control activities. During this phase, it’s important to identify and document individuals and teams that have an impact on medical device design.
A design control software system within a broader eQMS which also functions as document management software makes the design control planning phase much easier. These types of solutions allow design and quality teams to share, store, and search important documentation related to design control and quality assurance.
5. Consider your inputs
According to 21 CFR 820.3(a), a design input is described as the “physical and performance requirements of a device that are used as a basis for device design.” (ISO 13485:2016 7.3 covers much of the same design input requirements.)
When it boils down to it, there are many types of design inputs — including performance data, regulatory requirements, labeling/packaging, device sterilization, and more.
Both 21 CFR 820.30(f) and ISO 13485 mandate comprehensive documentation related to product realization (e.g., all design control and quality processes).
Here, too, design controls software can be a vital tool for alleviating some of the strain that design control documentation places on your product and quality teams.
6. Consider international differences
We've talked a lot about FDA design control guidance.
Design control ISO 13485 requirements are broadly similar, but not identical. You can compare the two strands with this handy chart:

Fortunately, with the 2024 approval of the QMSR FDA design control guidance, and the broader 21 CFR 820 guidance with it, is set to become more closely aligned with international ISO 13485 expectations.
This harmonization should make compliant design controls for medical devices easier to embed for international organizations in future.
Accelerate product development with design controls software
What is design control? It's critical for the integrity, safety and compliance of your medical device. Getting robust and holistic design controls in place is a difficult undertaking for even the most advanced quality control teams.
But dedicated design control software like Qualio can help you streamline the entire process. From document management to event tracking, our eQMS is designed to help teams develop a proactive approach to quality management and compliance.
And with audit-ready reporting, quality teams can easily generate documentation and reporting. These feature-rich reporting capabilities help save teams time during audits, since most auditors will want to see detailed reporting related to your design controls and inputs.
Qualio’s cloud-based design control software ships with FDA and ISO best practices baked in. As a result, teams can focus their efforts on product innovation instead of constantly responding to quality or compliance issues.
Design controls for medical devices can be complicated—let Qualio simplify it with software designed specifically for the medical device industry. Our industry experts created a platform that can help your medical device company navigate requirements, and we’ll also provide you with direct access to those experts who can give you valuable guidance.
Contact us today to set up a demo!