The ultimate guide to change control for life sciences companies
For any organization that makes a product, it's important to have change controls in place, but what are change controls? How do they differ from document and records control?
The defining factor that sets change control apart from document or records control is the context. Change control standards and requirements are always specified as part of the design and development process. Document and records controls are one factor of the larger change control, along with process controls, and other system controls. For manufacturing companies, the level of change controls necessary can vary widely depending on the type of product being manufactured and the applicable regulations.
The more risk introduced by a change, the more involved the change control process needs to be. This is your comprehensive guide to what that change control process should look like. We'll cover definitions, an overview of applicable standards and regulations, considerations for change control software, and provide a detailed explanation of the process with examples sprinkled throughout.
After reading this guide you should be prepared to start writing your own change control procedure for a change control process that is compliant and comprehensive.
What is change control?
Imagine for a moment that a manufacturer has jumped through all the design and development process hoops and has finally brought a product to the commercial market. A few months after the release of the commercial product there is a supply issue with one of the materials used in the product and the manufacturer needs to use an alternative material. Change control is the process for ensuring that the material change is properly documented, validated, and implemented.
When manufacturing any heavily regulated product, significant changes may be subject to regulatory review. For some changes there may be a required notification process either before the change is implemented or after the implementation. If the change is significant, there may be additional testing required to demonstrate that the drug or device is still equivalent to the initially approved version. The change control process ensures that all of these requirements are met and that the process is executed in an orderly manner with clear documentation of all of the steps that were taken. Although it seems like a lot, change control requirements exist to ensure devices and drugs continue to be safe and effective even as time goes on and changes are made.
Change control standards and regulations
Change control requirements are specified in a number of standards and regulations. The most basic starting point is ISO 9001, a general standard for quality management systems (QMS), but it includes specific requirements for making changes and specifies that all changes must be clearly identified, reviewed, and controlled. The more stringent ISO 13485, which focuses on QMS for medical device manufacturers goes further, to ensure that identified changes are reviewed, verified, validated, and approved.
Looking closer at the regulations for the life sciences industries, there are change control requirements embedded in 21 CFR Part 211 and EU Regulation No. 1252/2014. These requirements specify change control requirements that are specific to the pharmaceutical industry. For example, EU 1252 specifies that manufacturers must evaluate impacts that changes to the manufacturing process may have on the quality of the active substance before implementation. It also requires suppliers of active substances to notify anyone they supply if there is a change.
The following is a good starting list of regulations, standards, and guidance documents you may want to consult when developing your change control procedures to ensure you're meeting all of the requirements that apply to your drug or device. Both the FDA and the EMA have numerous guidance documents available, so be sure to check them out for specifics. This list is not exhaustive, so make sure to perform your own due diligence to verify all applicable requirements are met for compliance.
- International Organization for Standardization, Quality Management Systems - Requirements ISO 9001:2015
- International Organization for Standardization, Medical devices - Quality management systems – Requirements for regulatory purposes ISO 13485:2016
- FDA 21 CFR Part 11 Electronic Records; Electronic Signatures
- FDA 21 CFR Part 211 Current Good Manufacturing Practice for Finished Pharmaceuticals
- FDA 21 CFR Part 314 Supplements and other changes to an approved NDA
- FDA 21 CFR 807 Subpart E Premarket Notification Procedures
- FDA 21 CFR Part 820 Quality System Regulation
- European Medical Device Regulation 2017/745
- FDA Draft Guidance for Industry - Computer Software Assurance for Production and Quality System Software, September 2022
- FDA Guidance for Industry - Manufacturing Site Change Supplements: Content and Submission
- Commission Delegated Regulation (EU) No 1252/2014 of 28 May 2014 supplementing Directive 2001/83/EC of the European Parliament and of the Council with regard to principles and guidelines of good manufacturing practice for active substances for medicinal products for human use
- ICH Q9 Quality Risk Management
- ICH Q10 Pharmaceutical Quality System
- ICH Q11Development and Manufacture of Drug Substances (chemical entities and biotechnological/ biological entities)
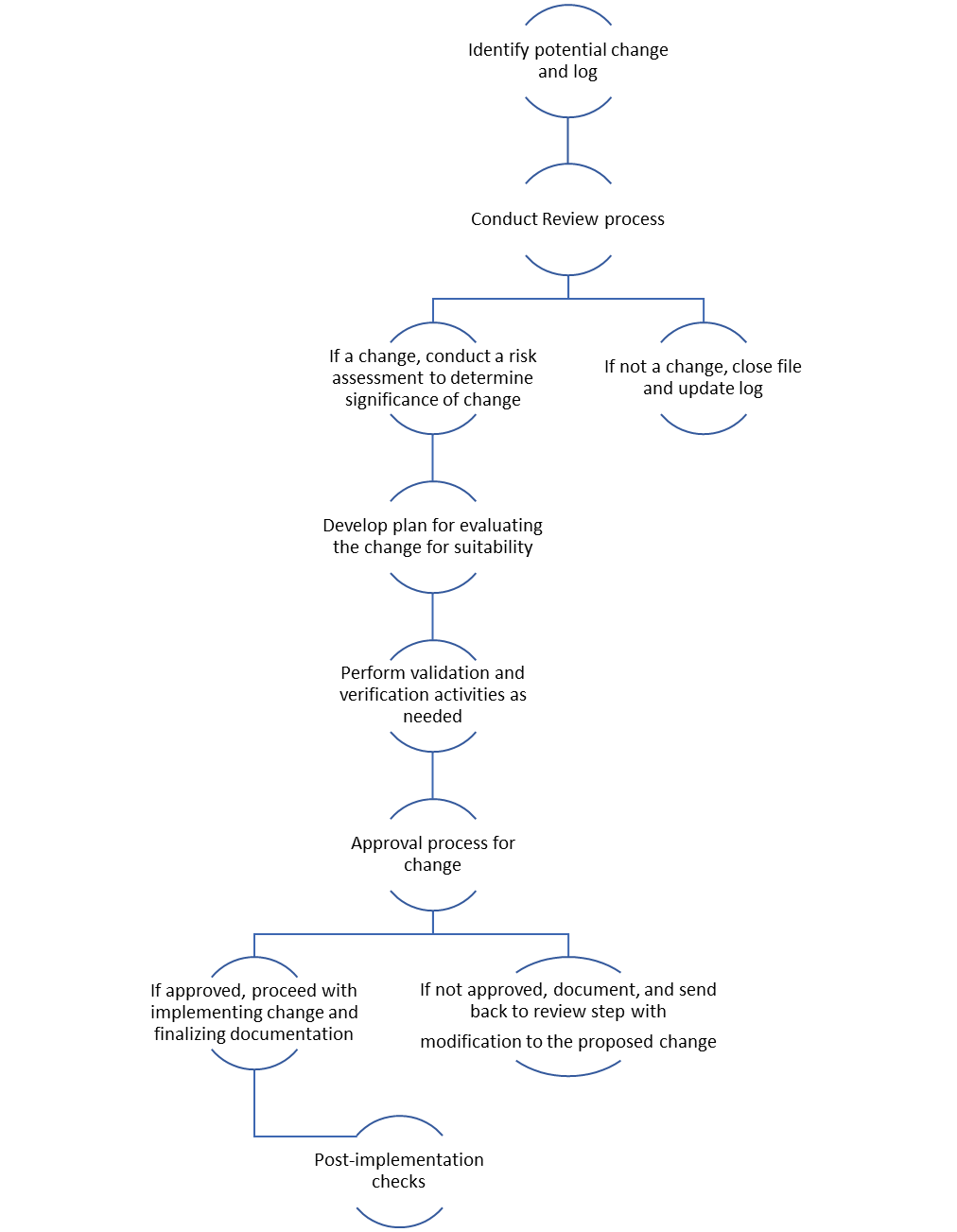
Change Control Process
1. Identify
What qualifies as a “change”?
Any change to a specification, method, process, or procedure of a released product must go through the change control process. Even seemingly minor changes such as updating a logo or the font on a label should be evaluated to determine what the potential impact will be.
Changing out a piece of machinery on the manufacturing line for a newer model is another example of a change that needs to be evaluated through the change control process. Changing process parameters for manufacturing the drug or device would be an example of a change that must be evaluated. This will then be further analyzed during the review process to determine the significance of the change based on how the change will affect the function, performance, usability, or safety.
All changes should be inputted into the change control system to begin the review process. If you are unsure if something qualifies, it's generally a best practice to err on the side of caution and send it through to the review process. If the change ends up not qualifying as a design or development change, then the review process will identify that fact and the change control file can be closed without further action.
Since this is the start of your process, any identified potential changes should be entered into the change control system at this point before proceeding to review. Once a potential change has been identified, the change will need to be entered into a log and some sort of identification number assigned.
If you're using a QMS software solution, it likely has a process built in to manage this. If you're using a manual process, you'll want to ensure that all of the steps for documenting the potential change are clearly specified in a procedure and/or a work instruction. The identification should include a description of the change and any other relevant or supporting information that will help the reviewers make a further determination about next steps and the plan for implementation.
2. Review
The design change review process should include a cross-functional team, much like the initial design and development process. The procedure or work instruction for the change control process should be developed with detailed instructions for how the review is to be conducted and documented.
Gather relevant documents and records
Prior to conducting the review, key documents and information about the proposed change will need to be assembled and provided to the review team. These documents may include things like material certifications, results from preliminary research and development (R&D) testing, and any rationale or other supporting documents for the change. If the change is being instituted due to a nonconformance or similar issue, all corresponding documentation should also be made available to the review team. All of this documentation will be used by the review team to determine the significance of the change and thereby the scope for verification and validation activities that will need to occur.
If you're using an automated system, the system will likely allow for these items to be attached to the change control record for easy dissemination to the review team, live commenting and discussion on the documents, and redlining as needed as the documents move through the change process.
Initiate change review
As part of developing a change control procedure, a change control review form or similar should be created to provide structure and guidance for the review process. This documentation should result in a concise summary of the documents and records reviewed, the team members that reviewed the change, and any plans that the review team develops for proceeding with the change control process. If you're evaluating a software solution, you will want to make sure that the system allows for a similar record to be created in the system that can easily be reviewed by an auditor.
A sample change control review form would include the following at a minimum:
- Identification of the proposed change
- Any supporting information about why the change is being made and the extent of the change
- Identification of the individuals performing the initial change review
- The results of the review
Conducting the review
One of the first priorities of the review team is to determine the significance of the change. The significance will be based on the potential risk that the change introduces. Even if a change is being made to reduce overall risk, the process of implementing the change will inherently introduce some risk. For example, even small modifications to a medical device may require updating the instructions for use, which then has a trickle-down effect with potential risk in ensuring that the correct revision of the instructions for use is included.
When identifying the significance of the change it is helpful to have a low, medium, high severity scale, or similar. A change that requires validation activities will usually fall in the high category. Examples of these type of major changes may include changing a piece of equipment or a process step. Changing the manufacturing location is considered a significant change and will also require working with regulators and the notified body, if applicable, to ensure that the new location is considered acceptable for manufacturing.
The results of the review may be inputted in a change control planning document. The basic template for this document may include a checklist of typical actions that need to be taken for a change to be implemented including: updating risk assessment documents, documenting all verification and/or validation activities, updating process and/or production documents, updating procedures, and updating regulatory documentation. This is not an exhaustive list but acts as a starting point for the review team to develop a plan. Another approach would be to develop an exhaustive list and then mark off items that apply or do not apply to a specific change instance.
3. Verification
The process for verification of a change will depend on the type of change, but in general it ensures that the planned inputs for the change meet the output. For example, if a dimension on packaging is changing, it would be verification that the packaging manufacturer is able to meet the new specifications. For a component, this would mean verifying that new dimensions are conformed to or that the component can be successfully made with a new material or in a new color.
Verification is NOT the same as validation. Verification is like a check to make sure that it can be done as planned for the change.
4. Validation
All validation activities must be planned and documented using an approved protocol. This protocol should be reviewed by personnel in the same roles as those approving the original validations. The validation must be executed under controlled conditions to ensure that the data is as accurate as possible. In general, protocols for validation of a product change will likely be very similar if not identical to the validation activities for the original product.
Validation can be a costly process, but for significant changes it is necessary to ensure that the manufactured product continues to be safe and effective. Validation for a change can hopefully be an abbreviated process compared to the initial validations conducted during development.
For example, if the change is only to a colorant it would not be expected that packaging validation would be required again. Further, it may be possible to just do a comparison of the two colorants to identify substantial equivalence in the verification step. That equivalence could then be used as a rationale to support that further validation activities are not required.
5. Approval
The approval process is the final piece of the process before the change can be implemented and approved for release. The approval process should look at all of the evidence from the review, verification, and validation steps. The approvers must include all of the people and/or people in the same role as the initial approvers of the original product. This means that you will again need a cross-functional team to approve. Ideally, the team should also include someone that is independent of the change control team. This independent reviewer acts as a fresh set of eyes. When a team is narrowly focused on getting a change through the system it is human nature to tend towards a sort of tunnel-vision, potentially missing other risks or issues that the change will introduce to the system.
Approval checklist:
- Has the risk assessment been updated?
- Have regulations been reviewed to ensure notifications have been made or are made immediately after this approval?
- Have all verification and validation activities been completed?
- Are documents, specifications, process documents, etc. ready to release after approval?
- Have all parts of the change control process been documented properly?
- Is there a clearly documented rationale for the change as part of the documentation?
The approval of the change will need to be documented, including the identities and roles of all approvers. If you are using an electronic signature process to document the approval, make sure that the signature is compliant with 21 CFR Part 11 and includes the date, time stamp, and the intent of the signature.
6. Notification
Regulatory authorities
For both medicinal products and devices there are regulatory statutes that determine notification requirements and processes. For medicinal products in the EU, there's Commission Regulation (EC) No 1234/2008 and the corresponding guideline that details how variations from the original marketing authorization are to be reported to the authorities. For both EU and FDA annual reporting is generally utilized for minor changes, while reporting prior to release of modified product is required for more significant changes.
For significant device changes, regulatory authorities may need to be notified of the change either prior to implementation or within 30 days of implementation. It may even be required to submit a new 510K for a device if the changes significantly affect the safety or effectiveness of the device. If you are using a notified body and the change is a significant change you will want to loop your notified body in near the end of the verification process and confirm what type of documentation they will require for the change, if any.
It's a good practice to develop a decision flow-chart that uses all regulations that apply to your specific device or drug to determine which notifications are necessary. This individualized flow-chart will allow you to ensure that you are considering notification requirements for all countries that your product is distributed to and the specific requirements for each national regulator. This document will need to be evaluated at least annually to determine if updates are needed, as regulations are ever-hanging. If you do develop a flow-chart, a copy of the decision process should be added to the change control file as a record.
Other notifications
When creating your notification decision flowchart, you should also include any other notification requirements for product registrations that you may have in various countries. For example, including things like the Unique Device Identifier (UDI) database update notification requirements would be useful. In a case where the manufacturer changed locations the UDI database will need to be updated to reflect that. Per the EU MDR, these updates must be made to the database within thirty (30) days of the change being implemented.
Changes to any information within the EU Single Registration Number (SRN) database must be submitted within one (1) week of the change being implemented. The list goes on an on and is highly specific to your device or medicinal product, so make sure that your regulatory team is considering all angles as part of change control.
Additionally, if you are supplying your product to any distributor or further manufacturer that may use them in their product, such as to create a procedure pack, you will need to provide them with a notification of the change and any supporting information requested. The type of changes that require notification and the level of detail required should be specified in an agreement with the customer.
7. Implementation
Once the change has been through the entire change control process and any regulatory authorities and your notified body have signed off on the change, if applicable, the change can be implemented.
When implementing the change, evidence of training for personnel and permanent implementation of the changes must be retained in the quality records. This implementation process may include putting process control documents or product specifications through the document control process. There may be changes to machinery that are needed to be permanently implemented, such as changing settings that were validated during the change control process. Training for personnel will need to be clearly documented. It is recommended that a verification of the effectiveness of that training is included as well. This may include some sort of testing on the new process through a simulation, actual production runs under supervision, or something similar.
8. Post-implementation activities
After the change has been implemented and new lots of product have been manufactured, it may be desirable to temporarily increase the quality checks depending on the type of change being implemented. For example, if the change is to the sterilization process, a higher level of testing may be warranted sporadically to increase confidence.
For a change to a manufacturing parameter like temperature or time, it may be desirable to increase the inspection level for in-process or finished product temporarily to ensure that there are no unforeseen adverse effects. Any of these post-implementation quality activities should be outlined as part of the change control plan. Think of this as being like an effectiveness check for a corrective action (CAPA). Records should be maintained for any post-implementation activities.
Additionally, the quality team should be on high alert after a change for trending in quality data. This is where unforeseen consequences of the change could appear. Any changes in risk levels should be added to risk documentation as appropriate.
One final best practice post-implementation activity is to have a wrap meeting to discuss lessons learned. This is particularly worthwhile for large, significant changes. This meeting can be used to improve the change control process and better prepare the team for future changes. Consider what parts of the process worked well and which seemed clumsy or chaotic. If there were issues during the final implementation, consider prevention of that issue in the future by adding a step earlier on in the change control process. This type of wrap meeting can result in great input for the preventive action side of your CAPA system.
Creating your change control procedure
If the product the company makes is a low-risk product that's not going to have a significant impact on safety, the change control process will not need to be substantial. For example, a medical device manufacturer that makes gloves would need a less robust change control procedure than a manufacturer of a pacemaker.
Any change control process should contain the same basic steps that were discussed above, but the level of detail needed in the procedure will probably be less for a lower risk product. The verification and validation steps will still be there, but you may not need to have different levels of verification and validation activities specified, for example. However, if the company makes a variety of products or it's expected that it may later expand into higher risk products, then it may make sense to spend the time now to write a detailed procedure.
What changes do not require change control?
Changes to quality system procedures that do not impact production do not require use of the change control system. These changes would be captured through document control and subject to review and approval in that system. Changes to processes that are business or quality based also would not require change control. For example, changing the accounting software and ERP system would not require change control, although this process would still need to be closely monitored to prevent business continuity issues with potential issues with ordering raw materials or fulfillment if the production flow is disrupted.
Making minor administrative corrections to specifications do not count as changes. For example, if there was a typo on a material called out in a specification this could be changed through document control alone and does not need to go through change control. The document control process would still capture an explanation of the change and the personnel that are reviewing and authorizing the change to be made.
If a manufacturer wants to be proactive and minimize supply chain risk later on, during the design and development process there could be alternative versions of a drug or device approved. For example, ordering a key compound from 2 different vendors during the development process would allow for verification and validation to be completed all at once. This would then allow for a quicker swap to the alternate version if a supply chain issue occurred. If the alternative was already approved, then the change control process would not need to be used since it would not be considered a change.
Managing supplier changes
Depending on the product, manufactured suppliers may or may not be required to report changes to the manufacturing process to their customers. For example, if a company manufactures an active drug substance and makes a change to the process, they must notify their customers per EU regulations. These or similar regulations do not generally cover all manufacturers of components, materials, etc.
To make sure that you're being informed of changes that may affect your finished product, you'll need to have quality agreements in place with your suppliers. A quality agreement is essentially a contract that is specific to product quality. This quality agreement should specify acceptable quality thresholds, the change notification process, record keeping guidelines, allowances for audits, and any other quality guidelines you need to control.
If a supplier notifies your organization of a change that change should then be evaluated to determine how it will impact your end product and if it continues to be acceptable. This should all be documented through the change control process.
Change control software
The change control process is complex with many moving, interconnected parts. Although the process can be executed manually with a detailed procedure and well-designed forms and tools, it may be beneficial to consider a software solution.
When looking into software for change control you will ideally want a system that is integrated and can facilitate pushing the process along with notifications and deadline management built in. A software solution is only beneficial if it automates pieces of the process. For example, after the initial review a team member may need to enter the various tasks into the system, but after that point the system will allow for management of deadlines, responsibilities, and routing documents and records to different personnel for review and approval as needed.
Change control software can seem expensive to smaller companies, but a cost-benefit analysis will likely show that software is cost effective, especially over the mid to long term. Consider if it is worth the risk of non-compliance or slow implementation of a change due to a manual process.
Things to look for:
- Customizable/configurable to meet your specific needs
- 21 CFR Part 11 compliant
- Integrated with other modules (i.e. document control, risk assessment, etc.)
- Automatic triggers to request approvals, alert personnel about deadlines, etc.
Software changes
There is increased focus on the role that software plays in the manufacture of drugs and devices both from a production standpoint and from a quality management standpoint. The FDA recently released a draft guidance document Computer Software Assurance for Production and Quality System Software. The draft guidance specifies the types of changes that are subject to comprehensive validation efforts.
In summary, QMS software or production software that could affect the safety of the device or drug need to be subject to significant validation processes, while all other software can be validated to a lesser extent, although it should be subject to a risk evaluation.
For example, software that controls process parameters such as temperature or process time needs to be validated to a higher degree since this could affect the safety of the drug or the device being manufactured. QMS software that automatically analyzes data to make decisions or release product should also be subject to the highest level of validation. For example, if an Excel spreadsheet or a similar software imports data from a temperature gauge and then automatically declares that the production range was acceptable, that would be considered critical to production. If the software imported the data but then was reviewed by a human, the software would not be critical to production.
Software changes can be captured through the change control system or through a separate software change evaluation system. For simplicity it may be easiest to embed software into the standard change control system. This would ensure that the cross-functional team is able to review software changes for possible wider impacts. Additionally, this contributes to a culture of quality and greater awareness of the need to potentially validate software. Excel spreadsheets are used ubiquitously in industry, but not everyone knows that in many cases they need to be properly validated and controlled in order to be in compliance.
Looking for change control software? Learn more about Qualio's eQMS.
Change control challenges
Implementation timing
Timing is everything in the change control process and it can be tricky to get just right for an effective and efficient process. Once a change finally makes its way through the approval process the team will want to pull the trigger and implement the change immediately, but that is easier said than done. Documents still need to be put through document control, production personnel still need to be trained, and equipment may need to be permanently installed on the line.
The key to trying to implement as quickly as possible is to stage as much as possible ahead of time. This means that Document Change Orders (DCOs) should be prepared ahead of time and ready to send out for final approval and sign-off once the change is approved. Alternatively, if your system is setup to allow it, the DCO can be already approved but not posted for live implementation until the change control approval is completed.
Training and equipment changes cannot be prepared ahead much, although any necessary training materials should be developed during the change control process both to ensure that training will be adequate and to speed implementation along. Equipment will need to be ordered ahead anyway for validation, any initial maintenance or setup activities can be completed if they do not interfere with an existing production line. For example, if the change under evaluation is adding a new production line, the new line can be fully setup ahead of time as long as it is not put into use until the change control process is complete.
Risk assessment
When conducting the risk analysis for a change that is being made, there will need to be a review of the existing risk assessment documents. It is easy to fall into the trap of simply evaluating the direct effects of the change.
When making a change there are inherent quality risks that should be taken into consideration as well. For example, if using a new vendor for a component, does the fact that it is a new vendor change any of the risk estimates since the vendor quality may not be as well known? Does the change to the process introduce new risk since personnel may be used to the old way? Additional mitigations that are taken as a result of the change should be added to the risk assessment documentation as well.
Just like in the initial development process, risk should be reviewed throughout the change control process as new information is received. For example, the risk assessment during the initial change review will have only enough information to determine if it makes sense to proceed to verification and validation. Once the verification and validation activities are completed, the risk assessment should again be reviewed using information from that process. New information may include process performance statistics, identification of potential quality issues, or even unexpected effects from the change under review.
Change control follow-up period
The change control process doesn’t stop immediately once the changes are implemented. There needs to be continued checks in place to ensure that the change was effective and that it did not create any additional issues.
Your quality team will most likely be responsible for handling this phase of the change control process. This follow-up period will provide important inputs for your risk management process and will help build a knowledge base for future change control implementation. This is not a step that should be skipped!
Key strategies for change control
- Having a strong change control process in place is critical for compliance.
- Developing a regulatory notification decision flowchart or matrix now can make your life much easier later.
- Don’t forget to document everything! You’ll need to document every piece of the change control process from identification through to approval and implementation.
- Using a software system for tracking the MANY moving pieces during a change control process can minimize chaos and ensure that steps are not skipped.
- Quality or engineering cannot complete the change control process in a silo, it is a team effort that functions best when it has input from everyone.
- Don’t forget the follow-up activities to make sure that the change was effective and didn’t introduce any other problems.
The change control process at a glance
Manage change with complete control. Get a look at Qualio here.
