Everything you need to know about the FDA 510(k) submission
What is an FDA 510(k) submission - and how do we complete one?
It's a question asked by many medical device companies in their earliest days. The FDA's 510(k) submission process is, in a nutshell, a clearance process which every company wanting to bring a medium-risk medical device to the American market must pass through.
Almost half of all medical devices used in the United States every day will have passed through the 510(k) route. There are about 3000 510(k) submissions a year, but 30% of 2022 submissions weren’t even accepted for initial review.
It's a long, complex process which can catch medical device quality and regulatory professionals out - so we've assembled everything you need to know about the FDA's 510(k) submission process right here.
Table of Contents
- What is an FDA 510(k) submission?
- The 510(k) submission and medical device classes
- What is a 510(k) predicate device?
- Who needs to submit a 510(k)?
- The three types of 510(k) submission
- The FDA 510(k) submission process
- Clearance!
- Top 8 submission mistakes
- 5 top tips
- FAQs
- Accelerate your FDA 510(k) submission
What is an FDA 510(k) submission?
The FDA's 510(k) submission process is, in short, a quality and compliance barrier designed to only let safe, effective medical devices onto the U.S. market, and into contact with American patients.
The focus of the 510(k) process is to prove something called 'substantial equivalence'. In other words, the aim of the game is to prove to the FDA that the medical device you want to bring to market is broadly similar to another device that's already on the market, known as a predicate device.
The logic of your 510(k) submission is clear: your device is like this predicate device, and here's how. And since this predicate device is already on the market and doing its job safely, your device must be safe and effective too!
At Qualio, we're big on making quality and compliance as simple as possible. With that in mind, here's a nice easy definition that gets at the heart of what an FDA 510(k) submission is:
510(k)
A technical file given to the FDA proving that your medical device is like another already on the market
Your 510(k) submission should:
1. Demonstrate your medical device is ‘substantially equivalent’ to a predicate already on the market
2. House detailed technical, safety and performance device/test information to show your device is safe and effective
3. Prove you have a robust medical device quality and risk management system
Now that's cleared up, let's dig a little deeper.
The 510(k) submission and medical device classes
The FDA classifies all medical devices marketed in the United States into three risk categories: Class I, low risk, up to Class III, high risk.

The 510(k) submission process is generally applied to so-called Class II devices, with a medium risk profile.
There are some caveats to this: in some very rare cases, Class I and III devices might require a 510(k) submission. But as a general rule of thumb, only Class II devices require one.
A 510(k) allows you to bring your Class II device to market without clinical trials, by proving the substantial predicate equivalence we've already discussed.
Medium-risk medical device requiring a 510(k) submission are those which come into sustained and significant patient contact.
Example devices include:
- Catheters
- Blood pressure cuffs
- Insulin pumps
- Endoscopes
- Electric wheelchairs
- Pregnancy test kits
- Syringes
- Blood transfusion kits
- Contact lenses
- Surgical gloves
- Absorbable sutures
The first step of any 510(k) submission process is to be sure that it applies to you, and that your device isn't exempt from the 510(k) process.
Giving yourself a clear understanding of when to submit a 510(k) vs. a Premarket Approval (PMA) is also an important task to tick off early.
What is a 510(k) predicate device?
A predicate device is the existing marketed device which your 510(k) submission should prove is substantially equivalent to your device.
The predicate device should have:
1. The same intended use as your device
2. Similar technology to that involved in the function and operation of your device
3. The same level of safety and efficacy as your device
Your FDA 510(k) submission should prove all three of these criteria. If your device is definitely Class II and there’s really no substantial equivalent at all - which can happen if you have a really innovative medium-risk device - you’ll have to go down the de novo route.
510(k) completion can only happen with a suitable predicate at the core.
And of course, substantial equivalence is a slightly loose term which doesn’t mean ‘identical’. A purely identical medical device would offer no competitive difference or advantage to your business - so there's a tricky balance to strike here. Use your best judgment.
Who needs to submit a 510(k)?
There are 4 groups of actors who would, depending on specific business circumstances, be responsible for completion of an FDA 510(k) submission.
a. American medical device manufacturers
The most common 510(k) submitters.
Making a Class II device in the US and bringing it to market? The quality/ regulatory lead in your business would complete your 510(k) submission as part of your go-to-market activities.
b. Representatives of non-US manufacturers
The second most common 510(k) submitting group are the appointed representatives of medical device manufacturers outside the U.S.
FDA rules state that non-US manufacturers wanting to market their device in the United States require a representative to submit their 510(k) on their behalf.
c. Specification developers
Designed and developed a medical device but outsourcing production to a CMO?
The 510(k) submission is still your responsibility, not the CMO's.
d. Repackers/relabellers
It's not very common, but in certain cases repackers and relabellers in a medical device logistical supply chain could be responsible for a 510(k) submission.
This typically only happens as a kind of update 'special' submission (more on that below) in the event of a significant process change, such as if significant labelling changes are made (like adding a new use or warning to a manual), or if significant repacking changes are made in a way that could affect the safety and integrity of the device (like in a sterilization operation, for instance).
The three types of 510(k) submission
There are three types of 510(k) route:
%20submission%20types.png?width=1758&height=570&name=510(k)%20submission%20types.png)
It’s more than likely that you’ll fall into the traditional route, which is what we’ll be covering in detail here.
Don’t get too excited by the possibility of an ‘abbreviated’ route: this option actually takes around twice as long as the traditional process for the FDA to review and doesn’t require any less work on your part.
It’s only performed where you think it’s easier to prove that your device meets certain regulatory standards or guidance than it is to prove equivalence to a predicate.
The FDA 510(k) submission process
Now we've covered the basics of the FDA 510(k), let's take a look at the submission process itself.
a. Finding your predicate device
Don't overthink this first step. Just Google your medical device type and see what comes up!
Find an existing business and device and compare:
- Intended use
- Design
- Materials
- Safety
- Labeling
- Performance considerations: engineering, sterility, compatibility, software validation, etc.
This should give you a broad feel for your device category and how other potential predicate devices work.
Once you've done this, head to the FDA's 510(k) database. Do the same again: type in your device type and see what comes up.
Here, for instance, I can see 316 potential predicates for my pregnancy test kit:
%20database.png?width=601&height=397&name=FDA%20510(k)%20database.png)
Homing in on a single predicate device, I can see everything I need to start my 510(k):
%20predicate%20device.png?width=521&height=351&name=510(k)%20predicate%20device.png)
I’ve found a predicate device, and I can even see that that device itself has its own predicate device and was found to be ‘substantially equivalent’ (SESE) to another marketed pregnancy test.
Crucially, I can see the three-lettered product code (LCX) for this entire category of devices. So I can always go back and search this code if I want to check the entire group of substantially equivalent devices.
Remember: the wrong predicate device can scupper your submission and send you back to Square 1. So do some careful consideration here, always ensuring that the ‘substantial equivalence’ threshold is being met.
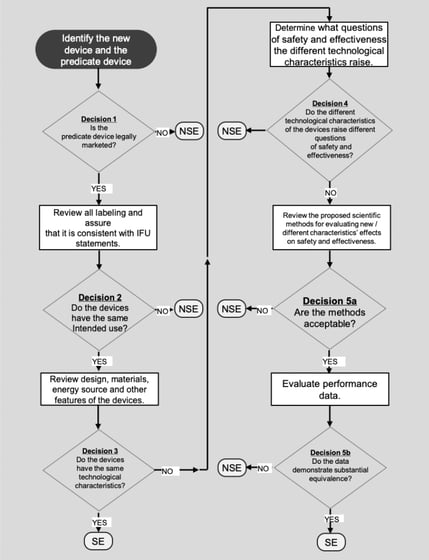
Once you’ve found a predicate with provable substantial equivalence, use these key ingredients to learn as much as you can about your predicate device:
- 510(k) submission summary
- A sample (probably lots of samples…)
- Diagrams
- Operating instructions
- Labels
- Marketing literature
b. Building a quality management system
An effective medical device quality management system with documented, standardized processes and procedures is crucial for underpinning your 510(k) submission and proving your device's safety and integrity.
Before submitting your 510(k), you should focus on embedding compliance with FDA 21 CFR 820, as well as ISO 13485.
Your QMS should house clear SOPs for all of the following areas:
- Regulatory and compliance strategy
- General safety and performance requirements (GSPR)
- Management responsibility
- Resource management
- Risk management
- Performance evaluation
- Product realization
- Unique Device Identification (UDI)
- Post-market surveillance
- Communication with competent authorities
- Incident reporting & field safety corrective action
- CAPA management
- Monitoring & measurement
Your QMS should also be supported by strong design, document and change control, with careful recording of your device lifecycle as the research, design and development stages unfold.
The more documentation you perform early, the easier your FDA 510(k) submission will be.
Learn more about the core requirements any medical device QMS needs for regulatory compliance
c. Device testing
Now we move onto the key 510(k) submission activity: performing your device tests to prove safety, efficacy and substantial equivalence.
The predicate device you chose in Step 1 will determine the tests you need to perform on your medical device. Your predicate is your benchmark and sets the acceptance criteria for your medical device testing.
Typical testing examples include:
- Mechanical/thermal/electrical
- Materials & make-up
- Wear/fatigue/durability
- Cleaning/sterilization
- Biocompatibility
- Electromagnetic compatibility
- Feature performance: normal and abnormal conditions
4 top testing tips
1. Use the product claims of your predicate device to set your test criteria
2. Use ISO 14971 as your benchmark
3. Look for historic recalls in your product category: use the faults as inspiration
4. Give yourself a digital mechanism for managing your design control activity
It's important that your business budgets wisely for this testing phase. Testing activity will form the bulk of your 510(k) expenses.
Get the right people and tools to maximize your in-house capability; typical third-party testing prices can quickly stack up:
- Third-party biocompatibility testing: $13,000
- Third-party sterilization testing: $15,000
- Third-party bench testing: $30,000
- Third-party electrical/EMC testing: $50,000
- Third-party implantation testing: $100,000+
If your device is an SaMD, you'll need to account for software validation activity too.
d. Submission
The FDA 510(k) submission itself is comprised of 20 key ingredients.
The FDA no longer requires a hard copy of the submission, and launched its Electronic Submission Template in September 2022 as part of a broad push towards digital submissions.
Following early success, in October 2022 the FDA announced its CDRH Customer Collaboration Portal would allow either an electronic copy (eCopy) or electronic Submission Template And Resource (eSTAR) 510(k) submission.
But now, after October 1, 2023, only the eSTAR is accepted. The eSTAR is a templated dynamic PDF you complete and submit electronically through the CDRH Portal (which you’ll need to sign up for). This more guided process is part of the FDA’s push to make 510(k)s faster and easier than ever before.
Let's dive into the key documents your 510(k) submission should include.
I. Cover sheets
The first two ingredients of your FDA 510(k) submission are your cover sheets:
- The FDA 3601 form (Medical Device User Fee Cover Sheet)
- The FDA 3514 form (CDRH Premarket Review Submission Cover Sheet)
The user fee cover sheet is your receipt for payment to enter the 510(k) process. The FDA website contains instructions for how to complete it.
The Premarket Review sheet is a basic summary of your submission, including company/device information, reason for submission, type of device, and so on.
Save $14,903 with a form
Make less than $100m annually?
Complete FDA Form 3602 (or 3602A for non-US firms) to register as a small business.
Small businesses pay just $4,967 for 510(k) submissions in 2023, compared to the $19,870 standard fee.
Businesses of all sizes also pay the annual establishment registration fee of $6,493.
Complete the Form 3602 every year to maintain small business status!
II. Summary documents
Next come your summary documents:
- Cover letter
- Statement of indications for use
- 510(k) summary OR statement
Your cover letter is a broad summary of your 510(k) submission.
Your indications of use statement MUST match the indications for use of your predicate device. It's made public after 30 days – so don’t reveal too much if your device is designed to fill a market niche!
For the final summary document, choose EITHER:
- Summary: combination of information that your SE claim is based on
- Statement: certification that you’ll provide safety/effectiveness information to any requester within 30 days
III. Statement documents
The 510(k) 'statement' documents are:
- Truthful & accurate statement
- Class III summary
- Financial certification/disclosure statement
- Declaration of conformity & summary reports
Not all of these documents may be relevant to you, depending on the circumstances of your submission.
The truthful and accurate statement is a standard, templated declaration as follows:
"I certify that, in my capacity as (the position held in company) of (company name), I believe to the best of my knowledge, that all data and information submitted in the premarket notification are truthful and accurate and that no material fact has been omitted. (Signature, date, time)."
As we’ve seen, most 510(k) submissions are for Class II devices - so the Class III summary is a simple declaration that you aren’t a Class III device. If yours is one of the rare Class III 510(k) submissions, you’ll need extra safety/effectiveness data here in line with your device’s higher risk profile.
The financial statements are only required if you conducted clinical trials for your medical device.
If you paid your investigators, complete the disclosure statement.
If you didn't, complete the financial certification.
And finally, the declaration of conformity & summary reports aren't required for the Traditional 510(k) route - they're for showing conformity with relevant standards in the Abbreviated route.
IV. SE documents
The SE documents, as you might expect, are for proving your device's substantial equivalence to its predicate.
We do this with three separate documents:
- Executive summary
- Device description
- Substantial equivalence comparison
The executive summary is a more detailed version of your 510(k) summary, elaborating on your completed testing activity.
Your device description should include your key technical information: Design History File, design outputs, drawings, specs, dimensions, and so on.
And your SE comparison is a side-by-side mapping of your device and its predicate, including the use, technology and performance of both devices.
V. Safety documents
The safety documents in your FDA 510(k) submission are:
- Proposed labeling
- Sterilization and shelf life
- Biocompatibility
Your labeling proposal should include your device label, patient labeling, in-package inserts and operating instructions. Your Unique Device Identification (UDI) code isn't mandatory at this stage, but you should include a sample barcode of where it will go.
If your device isn't sterilized or with a shelf life, you can skip the next section. If it is, include your test data for these areas here.
And for the biocompatibility section, include your test protocols and reports for any areas in patient contact (direct or indirect).
VI. Digital/electrical documents
If your device has a digital or electronic component, you'll need to include these next sections, Sections 16 and 17, in your 510(k) submission. If not, don't ignore them - briefly explain why they aren't relevant for you.
There are two documents to consider here:
- Software
- Electromagnetic compatibility/ electrical safety
For the software documentation, establish your device's Level of Concern (LOC) and add your test data accordingly.
Helpful software reference documents
Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices
Your EMC document should prove your device won’t interfere with, or be jeopardized by, other nearby devices.
The electrical safety document should prove that the electrical components in patient contact aren’t a threat to patient safety.
Helpful EMC/EC reference documents
It’s important to note that your device can still be a hardware device and include a software element. If your device falls here, you still need to account for all software operation in your 510(k) submission, even if you aren’t an ‘SaMD’ or 'SiMD' by definition.
Firmware-related software will still need documentation; your device's Level of Concern will determine what level of documentation is needed.
%20device%20software%20hardware.png?width=606&height=522&name=510(k)%20device%20software%20hardware.png)
VII. Performance documents
Finally, we arrive at your device's performance documents. These are arranged in 3 sections:
- Bench
- Animal
- Clinical
Only the first is mandatory; the other two may not apply to your operation.
All completed tests should have protocol descriptions, summaries, methodologies and clear results.
e. The wait
You've assembled the sections of your 510(k) submission.
You've gone over the FDA's Refuse to Accept policy to make sure you're meeting all of its 56 requirements and won't receive a refuse-to-accept letter.
Now it's time to press the button and wait.
How long does an FDA 510(k) submission review take?
The FDA's official timeline is as follows:
%20timeline.png?width=222&height=407&name=FDA%20510(k)%20timeline.png)
Despite the ‘ideal’ 100-day timeframe, the average approval time after submission is 175 days.
This is skewed by some excessively long device approvals however, with the median approval time standing at 85 days.
The average number of days it takes to clear a device via 510(k) also varies according to the device category.
Anesthesiology devices have the longest average length to approval, averaging 245 days.
Toxicology devices are the shortest on average, at just 163 days. In short, your business is looking at a 3-6-month waiting time after submission.
Clearance!
A medical device is never 'approved' by the FDA after a 510(k) submission.
Instead, you’ll receive a letter (no certificate) confirming your device is cleared for marketization in the United States.
You can now market your device, but must still comply with other requirements like:
- Registration and listing (21 CFR Part 807)
- Labeling (21 CFR Part 801)
- Reporting (21 CFR 803)
- GMP requirements (21 CFR Part 820)
Well done! Pour yourself a drink.
Top 8 510(k) submission mistakes
As we've already seen, a full 30% of 510(k) submissions in 2022 weren’t even accepted for initial review.
A right-first-time 510(k) approach gets you to market faster, eliminating wasted time and effort while generating revenue more quickly.
To help you, we've assembled the 8 most common mistakes made by medical device manufacturers in their FDA 510(k) submissions.
1. Incorrect templates/document versions
2. Choosing an unsuitable predicate
3. Inconsistent information in areas where information is repeated in the submission
4. Skipping non-applicable sections, instead of documenting why they aren’t applicable
5. Incomplete test information (summaries without protocols/reports)
6. Wasting money by not applying for small business status
7. Immature quality/risk management
8. Generally disorganized submission documents (no ToC, lax sectioning/numbering, etc.)
5 top tips
As an electronic quality management software provider, Qualio works with international medical device organizations every day. Our team is full of experts with decades of collective quality and regulatory experience.
To make your FDA 510(k) submission process even smoother, here are 5 top tips from the team.
1. Try and use a fairly recent device as a predicate. Devices older than a decade are frowned upon!
2. Ensure everything is covered - nearly two-thirds of submissions are slowed by Additional Information (AI) requests
3. The more design control, risk and quality documentation you can organically build as you develop your device, the faster and easier your 510(k) assembly will be
4. Substantial equivalence is everything! 15% of submissions fail because of the wrong predicate
5. Familiarize yourself with the eSTAR now, so you’re prepared for the October 2023 transition date
FAQs
We've hosted a number of webinars about the 510(k) submission process, and welcomed hundreds of international attendees through our virtual doors.
Frequently asked questions (FAQs) are a valuable way to pinpoint areas of uncertainty and confusion - so we made a note and answered them!
"Can a non-US manufacturer submit a 510(k), or does it need to be done by a US representative?"
Yes, a foreign manufacturer can submit their 510(k) application directly to the FDA. For convenience you could get the help of a US-based contact, but it isn't mandatory.
"What exactly is a 'reserved' Class I device?"
Reserved Class I devices are those 'special case' devices that the FDA believes meet the reserved criteria in section 206 of the Modernization Act and, therefore, would require a 510(k) under section 510(l). View a list of reserved devices here.
"Can you discuss the differences in the process when submitting a 510(k) to CBER as opposed to CDRH?"
Combination products are typically marketed under a marketing authorization type associated with whatever constituent part provides the primary mode of action (PMOA).
Combination products with a drug PMOA need a new drug application (NDA) or abbreviated new drug application (ANDA).
Those with a biologic PMOA need a biologic license application (BLA).
And those with a device PMOA need a PMA, de novo or 510(k) submission, depending on risk profile.
A single marketing application is generally sufficient for a combination product. In some cases, however, a sponsor may wish to submit separate marketing applications for different constituent parts of a combination product, and the FDA may consider this permissible.
"How can you see the labeling of a predicate device?"
The best way would to try to locate an example on the market. Labels for such devices are also commonly placed in the 'about' section of the manufacturers's app or website.
"If my Class II device is exempted from 510(k), what documents and/or reports are required to market it in the US? Do we need to inform the FDA before marketing?"
All medical devices, even if exempt, are required to follow the Quality System Regulation (QSR) mapped out in 21 CFR 820. In some cases, exempt devices may be exempt from Good Manufacturing Practice requirements under the QSR.
As for the specific documents/reports and the level of detail needed, this can vary based on your device and its intended use and claims.
"Are there any harmonized standards given like in the EU MDR?"
View the list of Recognized Consensus Standards on the FDA website here.
"I'm not sure how to determine if the predicate device we've found is appropriate for submission. What do I do?"
The FDA has a handy decision-making guidance document for this. View it here.
"Can we use a predicate device not marketed in the US?"
No, your predicate must be a US-marketed product.
"Can we use one of our own devices as a predicate?"
Yes you can, and lots of companies do! Just make sure it's substantially equivalent.
Accelerate your FDA 510(k) submission
Want a faster, more controlled 510(k) process?
Qualio provides electronic quality management software to help medical device manufacturers digitize, automate and strengthen their quality management activities.
Electronic documentation, controls, workflows and records simplify all aspects of your FDA 510(k) submission process and get you to market faster.
