21 CFR Part 820: the complete overview
FDA 21 CFR Part 820 constitutes the regulatory standard for the safety and quality of medical devices marketed in the United States.
It's simple: if you want to market your medical device in the US, you'll need to hit and maintain compliance with every requirement within 21 CFR Part 820.
21 CFR 820 is a complex, demanding standard which maps out the ingredients for a compliant medical device quality management system, and lays out how to manufacture safe and effective devices that work for your patients.
Here's everything you need to know to comply.
Table of Contents
- Introduction to 21 CFR 820
- Quality system regulation (QSR)
- 21 CFR 820 Subparts
- Subpart A - General Provisions
- Subpart B - Quality System Requirements
- Subpart C - Design Controls
- Subpart D - Document Controls
- Subpart E - Purchasing Controls
- Subpart F - Identification and Traceability
- Subpart G - Production and Process Controls
- Subpart H - Acceptance Activities
- Subpart I - Nonconforming Product Control
- Subpart J - Corrective and Preventive Action (CAPA)
- Subpart K - Labeling and Packaging Control
- Subpart L - Handling, Storage, Distribution, and Installation
- Subpart M - Records
- Subpart N - Servicing
- Subpart O - Statistical Techniques
- Compliance with 21 CFR 820
- Implementation of 21 CFR 820
- Role of 21 CFR 820 in ensuring medical device safety & performance
Introduction to 21 CFR 820
FDA 21 CFR 820 is shorthand for the Code of Federal Regulations Title 21, Part 820, which outlines the quality system requirements for medical device manufacturers in the United States. The FDA established the regulation in 1978 - with some revisions and updates along the way - to ensure that medical devices used on American patients are manufactured in a safe and effective manner.
21 CFR 820 does this by laying out the good manufacturing practice (GMP) expectations for medical devices, establishing requirements that manufacturers must comply with in order to ensure the quality and reliability of their products.
Compliance with 21 CFR 820 is essential and mandatory for manufacturers who intend to market their medical devices in the United States.
Overview of 21 CFR 820
The regulation, as you'd expect, touches all aspects of the medical device manufacturing process, which we'll dig into below.
These include design controls, document controls, purchasing controls, production and process controls, corrective and preventive actions, device labeling and quality system records. It also includes requirements related to complaint handling, servicing and installation activities.
In the FDA's own words, the 21 CFR Part 820 standard is designed to cover medical device:
"design, manufacture, packaging, labeling, storage, installation, and servicing..."
By adhering to FDA 21 CFR 820, medical device manufacturers are expected to establish and maintain a comprehensive quality management system that ensures the safety, effectiveness and reliability of their products.
Compliance with 21 CFR 820 is assessed through inspections conducted by the FDA, or authorized third-party organizations.
Importance of 21 CFR 820 in the medical device industry
By mandating a baseline level of quality system requirements for medical device manufacturers, 21 CFR Part 820 ensures that resultant medical devices are safe, high-quality and efficacious for their intended use.
Since medical devices, particularly higher-risk Class II and Class III devices, can impinge on human health in a number of ways, FDA 21 CFR 820 acts as a gatekeeping standard of quality that minimizes the risk of potentially deadly device faults.
This, in turn, maximizes public and patient trust, as well as the chance of successful application of devices for their intended usage.
21 CFR Part 820 has international significance too, mapping out elements of medical device quality assurance that are mirrored in other national legislation.
Because its a key national medical device regulation, 21 CFR 820 is often compared and contrasted with the international requirements of ISO 13485, particularly as American medical device manufacturers look to expand into overseas markets with their existing quality management systems.
The ISO 13485 vs 21 CFR 820 binary and the difference between the standards has made life difficult in recent years, but the FDA's upcoming QMSR aims to harmonize Part 820 with ISO 13485 in 2026, bringing American medical device best practice closer in line with international expectations.
Quality System Regulation (QSR)
21 CFR 820 and the FDA's so-called Quality System Regulation, or 'QSR', are really one and the same.
As a result, they're often used interchangeably: 'FDA QSR 21 CFR 820' is a frequent search term that's technically correct.
The QSR pertains to the current good manufacturing practices (cGMP) mapped out for medical devices within 21 CFR 820.
The different names for the same set of requirements aren't just a case of semantics - they underline the fact that 21 CFR Part 820 is ultimately about regulating the quality systems that shape how medical devices are manufactured and distributed to American patients.
Purpose of QSR
A quality management system is designed to ensure that the products and services that your business offers its customers fulfil requirements.
If you're a medical device company, that alignment with customer (or in this case, patient) needs ultimately means offering a device that does what it's supposed to, without posing unnecessary risk.
The QSR roadmap outlined in 21 CFR 820, then, is designed to give your organization a structured, repeatable, process-based mechanism for consistently making devices that work.
In turn, your business enjoys:
- Reputation, trust and business: your device works!
- Lower costs: processes are scrutinized to minimize wastage and maximize leanness and efficiency
- Lower risk: device risks are proactively planned for and mitigated
- Consistency: your entire team knows what to do, how, and when
- Product-market fit: user needs and requirements are fed into your device's very being with design controls, ensuring it satisfies them
21 CFR 820 Subparts
What does 21 CFR 820 actually say, then?
The regulation contains 15 sections, or 'subparts'. You'll need to understand and apply the requirements in all of them to embed lasting 21 CFR Part 820 compliance.
Let's look at each in turn.
Subpart A - General Provisions
This section provides over 30 definitions of key terms in the standard, then lays out its general provisions, including scope, exemptions and applicability.
It's an 'orientation' subpart, designed to define exactly what is to be achieved.
Subpart B - Quality System Requirements
This subpart outlines the core requirements for establishing and maintaining your medical device quality system.
Its three sections cover management responsibility, audits and personnel, respectively.
It covers areas such as the quality policy, your organizational structure, quality planning and management review, as well as building an internal audit cadence and ensuring employee competence.
FURTHER READING: What is a quality management system?
Subpart C - Design Controls
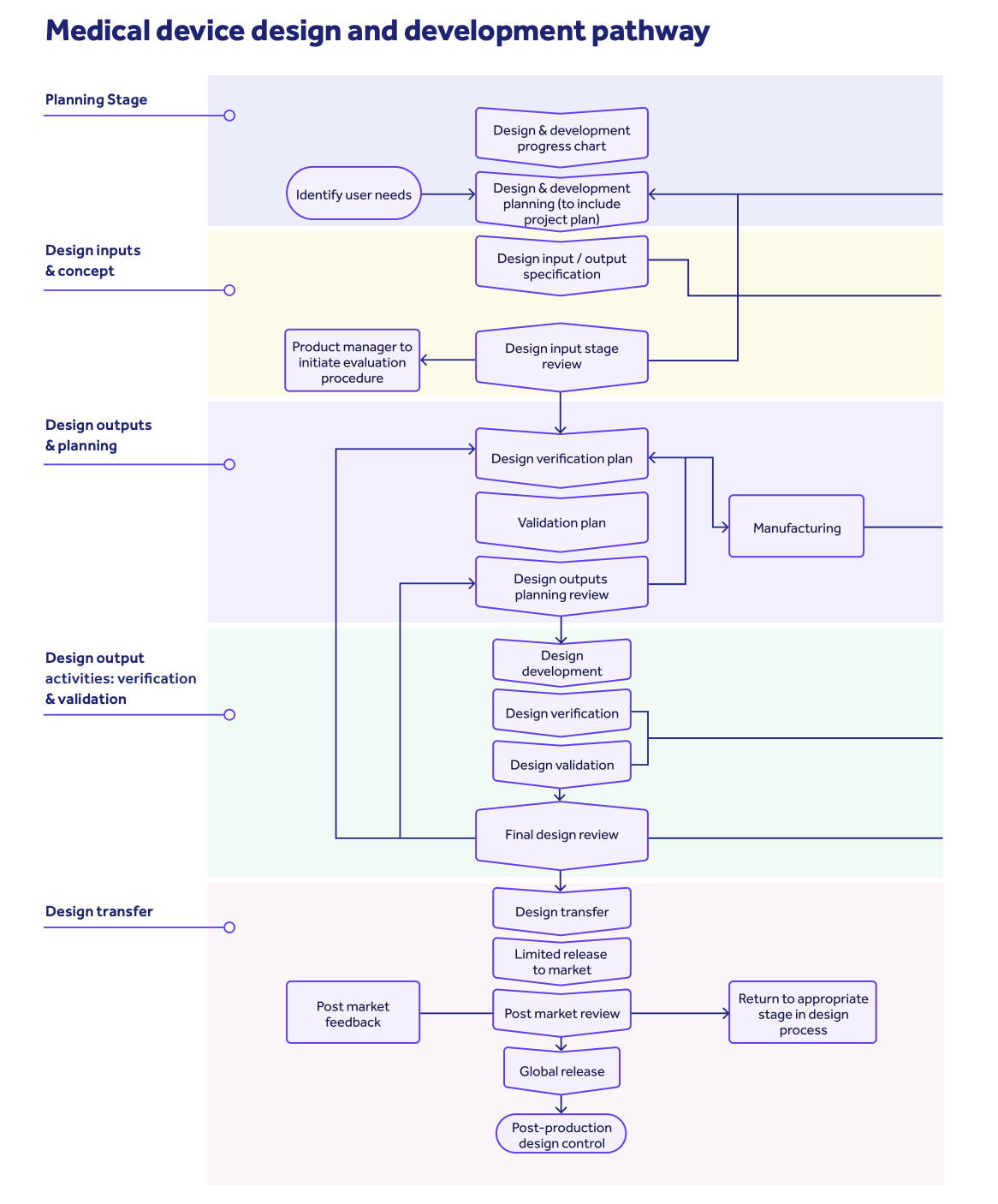
This section focuses specifically on design controls for medical devices. It includes requirements related to the key design control milestones of design and development planning, design inputs, outputs, verification, validation, transfer, changes, and the construction of your design history file.
Design controls, in a nutshell, are the practices required by the FDA to facilitate a structured design and development process for your medical device.
There should be multiple checkpoints in place to ensure your device is safe and effective when brought to market, as well as ensuring that user needs and requirements are properly integrated into your product design.
User needs should translate into design inputs, which then become realized design outputs which are then tested with verification and validation activity.
Subpart D - Document Controls
Robust quality management is impossible without clearly defined and controlled information streams.
This subpart details the requirements for document controls to meet this aim, including document approval, distribution, changes and retention.
FURTHER READING: 5 medical device document management tips
Subpart E - Purchasing Controls
The quality of your medical device is directly affected by the products and services that flow into your business from your supply chain.
This section addresses the controls and responsibilities for purchasing and accepting materials, components and services used in the manufacturing process of your medical device.
It covers supplier evaluation, selection, collection and usage of purchasing data, and verification of purchased products from your supply chain.
FURTHER READING: How to clear the 21 CFR 820.50 hurdle in 7 easy steps
Subpart F - Identification and Traceability
This subpart provides requirements for both identifying and tracing medical devices and their components.
The identification section focuses on giving your business the ability to pinpoint products during 'receipt, production, distribution, and installation'.
The traceability section concerns itself with being able to trace lots, batches and individual units of finished devices once they've left your four walls.
Subpart G - Production and Process Controls
This section outlines the controls and procedures for your production and manufacturing processes.
It covers various aspects, including equipment calibration and maintenance, inspection and testing, environmental and contamination control, personnel training and more.
Key elements to think about for Subpart G compliance are:
- Validation of computerized systems connected to your device design, development and manufacture
- Equipment
- Staff
- Buildings
- Materials
- Processes
- Change controls
Subpart H - Acceptance Activities
This subpart describes the requirements for your acceptance activities - in other words, how your business reviews and accepts incoming products.
Robust 21 CFR 820 acceptance activities include defining your acceptance criteria, then performing inspections, tests and other verification activities to ensure they meet your specified requirements.
Subpart I - Nonconforming Product
Quality management, as we've seen, requires consistent output of product which meet customer and patient demand. Nonconforming product slipping through the cracks should therefore be properly dealt with, and it's here that 21 CFR Part 820 Subpart I focuses.
This section addresses the controls and procedures for identifying, segregating, evaluating and disposing of any nonconforming products.
It includes provisions for responsibility assignment, investigation, segregation, disposition, and any required corrective actions related to nonconforming products.
Which brings us nicely to...
Subpart J - Corrective and Preventive Actions (CAPAs)
This subpart outlines the requirements for establishing and executing a corrective and preventive action system.
CAPAs are 'response' actions used to address a nonconformance in your medical device or any adjoining processes.
Nonconformances should be pinpointed, its causes assessed, and then tackled with targeted corrective and preventive action to stop it reoccurring.
CAPAs should then be measured and assessed post-execution, to ensure they've been effective.
NOTE: The CAPA requirements of FDA 21 CFR 820.100(a) were the most common cause of 21 CFR 820 non-conformances discovered in 2022! More on that below!
Subpart K - Labeling and Packaging Control
This section focuses on the controls and procedures for labeling and packaging of medical devices.
It includes requirements related to labeling accuracy and integrity, content and control, as well as procedures for device packaging and storage.
Subpart L - Handling, Storage, Distribution, Installation
This subpart covers, in four discrete sections, your requirements for the handling, storage, distribution and installation of your medical device.
It includes provisions for proper storage conditions, how to handle and distribute your device, prevention of mix-ups, contamination and damage, as well as installation controls for any devices that require installation.
Subpart M - Records
This section outlines the requirements for record keeping, including the types of records that must be maintained, their retention periods and the procedures for their storage and retrieval.
Two key components of Subpart M are the device master record and the device history record.
Subpart N - Servicing
This subpart addresses the requirements for servicing of medical devices. Not every device will need servicing, so check if this subpart applies to your operation.
Subpart N mandates procedures for servicing, maintenance, and repair activities, as well as requirements for servicing records and response to any complaint records.
Subpart O - Statistical Techniques
This section provides guidance on the use of statistical techniques for establishing and maintaining both process control and product quality in your medical device manufacturing processes.
Because quality management requires adherence with defined and measurable outputs, Subpart O encourages compliant statistical techniques to help you measure and verify success.
Compliance with 21 CFR 820
Now that we've covered the content of the standard, let's look at what 21 CFR 820 compliance actually involves and requires.
Inspection and observation process
Since FDA 21 CFR 820 is a federally mandated standard, medical device manufacturers marketing devices in the US will have their quality systems inspected by the FDA's Office of Regulatory Affairs (ORA) to ensure compliance with the requirements of 21 CFR 820.
The inspection process typically involves the following key steps:
-
Notification (or not!)
Generally, the FDA will let you know an inspection is coming around 5 days ahead of time and request some documents to review early. But in some cases, surprise unannounced inspections can occur - making constant audit readiness crucial! Inspections can be triggered by routine cadences, by a complaint, or by reported issues or faults with your device(s)
-
Opening conference
At the start of the inspection, an opening conference is held to introduce the inspection team, discuss the purpose of the inspection, outline the scope and objectives, and present you with your formal Form 482 'notice of inspection'
-
Document review
Your FDA inspector will then begin to apply the so-called Quality System Inspection Technique (QSIT), starting by examining various documents and records related to your 21 CFR 820 medical device quality management system, such as your standard operating procedures, device master records, design history files, complaint files and corrective and preventive action records
-
Facility audit
Your inspector will conduct a walkthrough of your manufacturing facility to assess the adequacy of your physical infrastructure, equipment and processes. They may also observe manufacturing activities, take samples, or interview your personnel to ensure they understand and follow your established quality system, and that your teams are adequately trained to execute their roles properly
-
Findings
Throughout the inspection, your inspector will document any non-conformances, observations and violations of your FDA QSR 21 CFR 820 requirements. We'll look at the most common violations below
-
Exit meeting
Once your inspection is completed, an exit meeting is conducted to discuss any findings and observations you've accrued. This is your opportunity to address any concerns or provide additional information. You'll then be given an overall inspection grade: No Action Indicated (NAI), Voluntary Action Indicated (VAI) or Official Action Indicated (OAI)
-
Observations or warnings
A VAI will result in the issuing of an inspectional observation (also known as a Form 483) while, in more serious circumstances, an OAI will see you get both a 483 and a warning letter. You'll be expected to respond to your noted deficiencies with a corrective action plan, which will be checked in a follow-up inspection. If you get an OAI, your next 2 years of inspections will be unannounced
-
Follow-up and enforcement
If you got an NAI, great! Maintain compliance and stay ready for any future FDA drop-ins.
But if you did, you should respond to your 483 or warning letter as soon as possible, and ideally within one month. The FDA will evaluate your response and determine if you've made sufficient corrective action to embed full 21 CFR Part 820 compliance. Ongoing non-compliance, obviously, will make things worse, and could result in more warning letters, in product recalls, or, in the most extreme cases, in complete shut-down of your organization
FURTHER READING: What should I expect during an inspection?
It's important to note that your inspection and observation landscape is largely shaped by the nature, risk profile and class of your device.
Class II and Class III manufacturers can expect biennial on-site inspections, while only a few higher-risk Class I manufacturers may receive an on-site check-up at all, since they aren't subject to some cGMP requirements.
It's also important to reiterate here that your 21 CFR 820 compliance focuses on your quality system.
Your medical device itself may also require additional FDA overview and inspection as part of a submission pathway, depending on its class:
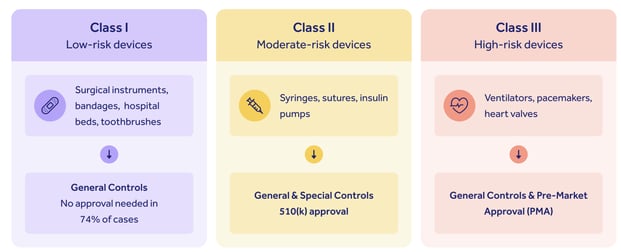
Medium-risk Class II devices will generally require a 510(k) submission, while high-risk Class IIIs need pre-market approval.
Think of your 21 CFR 820 compliance and your device submission pathway as two interacting halves of the same coin: you don't need to evidence your medical device quality system in a 510(k) submission, for instance, but having a robust 820-compliant QMS will give you accurate, reliable device data to feed into your submission pack.
Equally, getting robust, well-documented design controls and risk management processes in place will help your device sail through a submission and make your 21 CFR 820 inspector happy when they come to check up on your quality system.
A strong, functional quality system should be the cornerstone of your entire operation, whatever your risk class.
Common violations and non-conformances
Understanding the most common triggers of FDA 21 CFR 820 non-conformance, particularly of Form 483 inspectional observation issuances, is a good way to direct your efforts and avoid making typical mistakes made by other medical device companies.
See the 6 most common triggers for medical device Form 483 observations here
To view the entire list of inspectional observation trends, visit the FDA's website.
Effects of non-compliance on medical device manufacturers
We touched on what happens if you slip up on your FDA 21 CFR Part 820 requirements above.
Minor non-compliance will bring a 'Voluntary Action Indicated' and that Form 483 list of inspectional observations to address.
Major breach of 21 CFR 820 expectations will bring an 'Official Action Indicated' and a warning letter.
It's here that the effects of non-compliance on medical device manufacturers can be serious.
Alongside the effects of reputational damage and erosion of public trust, recalls, seizures, injunctions, civil action, financial penalties and even prosecutions and shut-downs are all in the realm of possibility for serious, protracted, consistent flouting of 21 CFR 820 requirements.
With all that in mind, let's examine some best practices and indispensable ingredients you'll want to get in place to embed airtight, lasting 21 CFR 820 compliance.
Implementation of 21 CFR 820
Securing compliance with FDA 21 CFR Part 820 requires three primary overarching ingredients, which we'll explore now.
Establishing and maintaining a quality system
Obviously, the Quality System Regulation's primary requirement is the establishment of a functional quality management system.
It's here that your 21 CFR 820 compliance journey should begin.
Without a QMS in place, it'll be impossible for you to guarantee the safety, quality and integrity of your device and pass FDA inspection.
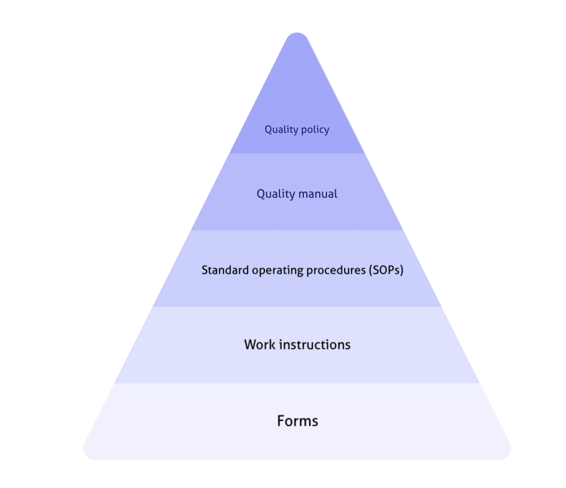
A clearly defined quality policy and manual should shape and combine all your operational quality ingredients, like SOPs and work instructions, like so:
And you should ensure your QMS fully defines, includes and controls the 21 CFR 820 requirements we've explored, like:
Start small, considering each requirement of both your device's end user and stakeholders like senior management and the FDA, then let those requirements translate into your QMS operating procedures.
Focus on your core processes first. Weed them out by asking yourself which processes and activities play the most significant role in ensuring your device meets customer, statutory and regulatory requirements.
This will give you a 'priority process' list, which should include elements like:
- Product design and development
- Risk management
- Incident management
- Documentation
- Feedback
- New supplier
- Equipment management
- Customer onboarding
- Internal auditing
- Data retention
- Competence management
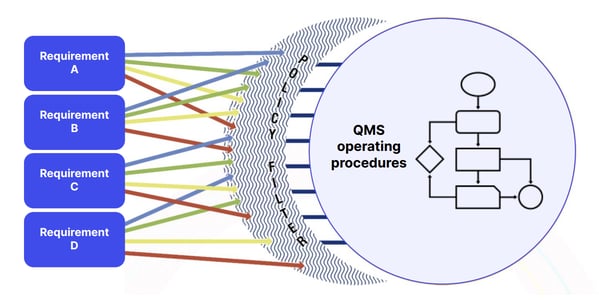
Once you've established the bare bones of your medical device quality management processes, it's a good idea to apply the 'As-Is' methodology to pinpoint any compliance gaps and ensure you're ticking off 21 CFR 820 requirements:
1) Determine how current processes are executed
2) Find areas of variation and non-conformance with internal audits
3) Learn what's working well and what is not
4) Ask yourself how your processes can be standardized and aligned with 21 CFR 820 expectations: how can steps, systems and tasks be integrated and improved, or, in some cases, combined or removed?
5) Make a tactical 'to do' list of SOPs, policies, instructions and so on which need to be created, distributed and trained on
DOWNLOAD YOUR GUIDE:
9 ways to improve quality in medical device product development
Integrating design controls and risk management
Once you have your overarching QMS in place, it's essential for your 21 CFR Part 820 compliance that you can demonstrate consistent embedding of design controls and risk management into the very heart of your device's lifecycle:
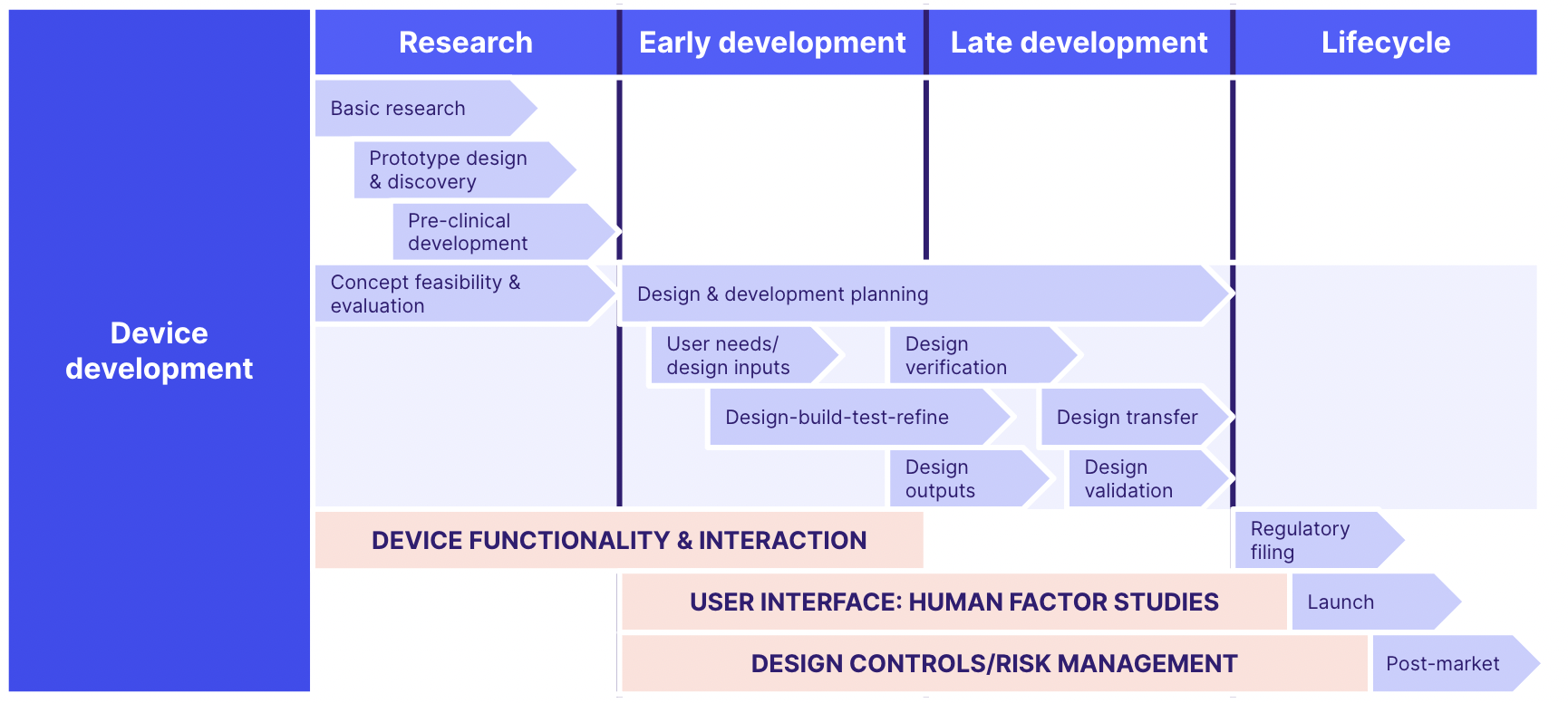
And for maximum effectiveness, your design controls should integrate with inputs from and outputs to your wider QMS processes, as follows:
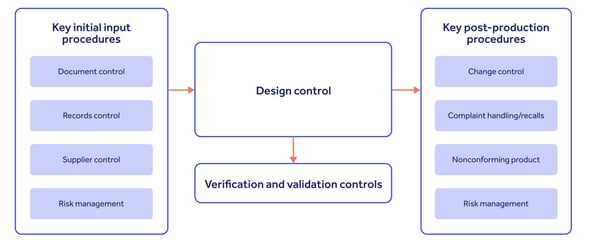
Don't be afraid to look to other sources for risk management best practice.
Some risk management frameworks to consider and apply to your medical device QMS include:
- ISO 31000/31010
- ISO 14971 (the medical device risk management standard)
- Failure mode and effect analysis (FMEA)
- HAZOP
- Cause and effect analysis
- Root cause analysis
- Risk indices
- Cost/benefit analysis
DOWNLOAD YOUR GUIDE: Design controls for medical devices: 6 principles for success
Developing a CAPA system
Getting an effective CAPA system in place isn't just about complying with Subpart J.
Robust CAPAs will touch all areas of your QMS: the stronger your CAPA processes, the more quickly and effectively you'll be able to stamp out defects, faults and weaknesses in both your device and its adjoining processes.
Things will inevitably go wrong at some stage as you design and build a medical device for the US market. Being able to efficiently spot, target and permanently fix errors is the difference between a good medical device company and a great one.
Role of 21 CFR 820 in ensuring medical device safety and performance
FDA 21 CFR Part 820 is there to help you ensure your medical device performs safely and effectively for your customers in the United States.
As we've seen, it's a complex, but necessary, standard for you to align your medical device quality management system with.
It's important that you treat your 21 CFR 820 requirements not as a hoop to jump through, but an opportunity to construct a robust, effective and quality-first approach that guides everything to do with your device.
Like any standard, 21 CFR Part 820 is constantly evolving and changing. The FDA plans to integrate it more closely with ISO 13485, the international medical device quality management standard, in the near future - so you should take a look at the areas of crossover and build your QMS accordingly to stay ahead of future trends.
It's crucial, too, that your team focuses on making 21 CFR Part 820 compliance as natural, easy, automatic and 'business as usual' as possible.
Legacy quality management tools like paper and spreadsheets may suffice in the earliest days of your operation, but will begin to creak, clutter and complicate your QMS as you near marketization and FDA inspection.
Quality-centric and forward-thinking medical device companies are increasingly turning to quality management software to accelerate and automate key quality processes like document control, training and CAPA execution. By doing so, FDA clearance and long-term medical device market success become simple.
Book a demo with Qualio to learn how we empower our customers to make FDA 21 CFR 820 compliance a natural byproduct of a world-class quality approach.
