What is ICH E6 R3? Good clinical practice explained
ICH E6 R3, also known as ICH E6 (R3), is a regulatory guideline mapping out the requirements of modern good clinical practice (GCP).
ICH E6 R3 is aimed at organizations designing and executing clinical trials, and is intended to help these clinical bodies embed repeatable and dependable quality into the entire end-to-end clinical operation.
If you want to provide a quality-centric, reputable and trusted clinical framework, ICH E6 R3's GCP guidelines are your starting point. Let's take a look at them.
Table of Contents
Introduction to ICH E6 R3
ICH E6 R3 is a guideline for good clinical practice developed by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH).
It's an internationally recognized standard that provides guidance on the design, conduct, monitoring, recording, analysis and reporting of clinical trials to ensure 'good' practice is adhered to.
Like all ICH guidelines, ICH E6 R3 isn't a binding, mandatory requirement that you have to follow. The US FDA states that 'an alternative approach may be used' and maps out its own GCP touchpoints in FDA 21 CFR Parts 50, 54, 56, 312 and 314.
But ICH E6 R3 is widely adopted and recognized by regulatory authorities, contract research organizations and other clinical research stakeholders to ensure the conduct of high-quality clinical trials and the generation of reliable, trustworthy data for evaluating the safety and efficacy of new medical products.
It therefore offers a ready-made, internationally adopted framework that positions your organization for multinational best practice.
To get robust GCP in place, ICH E6 R3 is your best bet.
Overview of ICH E6 R3
To understand the ICH GCP E6 (R3) guideline, it's important to get an overview of what good clinical practice entails and what the ICH wants to achieve with the guidelines.
GCP guidelines are designed to ensure that clinical trials are performed in a dependable, ethical way that aligns with the World Medical Association's Declaration of Helsinki.
Subject rights, safety, privacy and wellbeing should be maximized above all else - even the interests of 'science' and 'society' - while clinical trials themselves should be anchored on scientifically sound, risk-benefit principles that generate dependable, trustworthy data conclusions.
GCP, enshrined in ICH E6 R3, should set a shared, mutually accountable quality framework for sponsors, IRBs, investigators (CROs) and regulators to follow throughout the clinical process.
"Well-designed and conducted clinical trials help
answer key questions in healthcare and drug development. Their results are essential for evidence-based healthcare decisions.Trials with inadequate design and/or poorly conducted
trials may place participant safety at risk, yield inadequate or unreliable results and are unethical.They waste resources and the efforts and time of investigators and participant..."
- ICH E6 R3 introduction
Above all else, the focus of GCP and ICH E6 R3 should be seen as the pursuit of both subject rights and dependable clinical data together.
Scope
The guideline itself states that it applies to 'interventional clinical trials of investigational products that are intended to be submitted to regulatory authorities'.
And even for clinical trials that aren't connected to marketing authorization, the guideline could still be applicable.
In short, E6 (R3) intends to offer a shared GCP framework connecting sponsors, IRBs, investigators and their regulators from clinical trial design through to final data analysis.
If you're conducting any kind of interventional (clinical) trial, ICH E6 R3 will be applicable to you.
If you're operating an observational trial, it won't be.
History and evolution of ICH E6
ICH E6 R3 is an updated version of the original ICH E6 guideline, first published in 1996, and the ICH E6 R2 revision released in late 2016.
The 'R3' in the title, like in ICH Q8, signifies that it's the third version of the guideline, published after two revisions and updates of the original.
The ICH E6 (R2) integrated addendum was rolled out in 2016 with the aim of promoting technology and new innovative approaches within clinical trials, while also going further to ensure the protection of subjects and the reliability and quality of clinical data.
In short, ICH E6 R2 added emphasis in a few new areas:
1) The application of a risk-based approach across the design, monitoring and reporting activities of a clinical trial
2) The application of a QMS
3) New responsibilities for clinical sponsors
4) The use and management of electronic records
5) Monitoring and oversight expectations
We'll touch on these changes in more detail below.
Purpose of ICH E6 R3 guidelines
ICH E6 went live in 1996.
E6 (R2) replaced it in 2016, adding a new emphasis on both risk and quality management.
And E6 (R3) aimed to move the needle again, updating and evolving the ICH's expectations for how modern clinical trials are operated.
The evolution of the E6 guidelines has run in parallel with the broader changes taking place in the clinical sphere, such as the adoption of new technology and the cross-fertilization of other ICH concepts from the pharma world, like quality by design (QbD).
All three iterations have good clinical practice (GCP) at their core.
The protection of subject rights and safety, married with the output of robust, trustworthy clinical data, is the key objective of GCP, and both sponsors and investigators have their own set of responsibilities within ICH's E6 GCP framework.
Only about a third of FDA-regulated drugs make it to the critical Phase 3 of their clinical trial pathway: clinical trials are the ultimate test for a nascent drug or device, acting as the gatekeeper of public safety and a critical hoop for lifesaving products to jump through.
GCP, manifested in high-quality clinical processes, trustworthy data, airtight data integrity and clear roles and responsibilities is crucial for clinical trials to function properly. Like any subset of GxP, it sets the benchmark of acceptable practice for regulated companies to meet.
The ICH GCP E6 (R3) guideline is designed to let you embed GCP across all layers of your clinical operation:

As a result, your clinical trials are conducted with integrity, your data conclusions are reliable, and - most importantly of all - your patients and participants are properly protected across the entire experience.
Download our guide to clinical trial quality by design (QbD)
Key changes from ICH E6 to ICH E6 R2
To understand ICH E6 R3, it's useful to dig into what changed in the twenty years between ICH E6 and ICH E6 (R2).
Like any life science quality guideline, ICH E6 is open to change and reinvention as technology, regulations, expectations and objectives evolve.
The rollout of ICH E6 R2, followed by ICH E6 R3, shows that evolution in process.
ICH E6 R2 made 5 key changes to the original ICH E6 roadmap.
Risk management and quality control
Other ICH standards like Q9 place emphasis on quality risk management as an integral component of modern pharmaceutical quality systems.
Revision 2 of ICH E6 extended this focus into the clinical sphere, prescribing a double mixture of risk and quality management actioned through a functional quality management system (QMS).
Key elements of a clinical QMS reflected in Qualio's eQMS software
Clinical sponsors should start by identifying their critical data, and the processes it flows through, as protocols and study materials are built and developed.
Associated risks should then be pinpointed at both the operational level (QMS, SOPs, organizational structure, systems, processes, etc.) and then the tactical project level: from trial design to population, site, the nature of the investigational product (IP), data management, and so on.
WATCH OUR WEBINAR: Managing risks and opportunities in your QMS
Once these risk and quality factors have been found, Section 5 mandates that a demonstrable risk management strategy be established, encapsulating analysis, evaluation, and then measurable treatment and reporting:
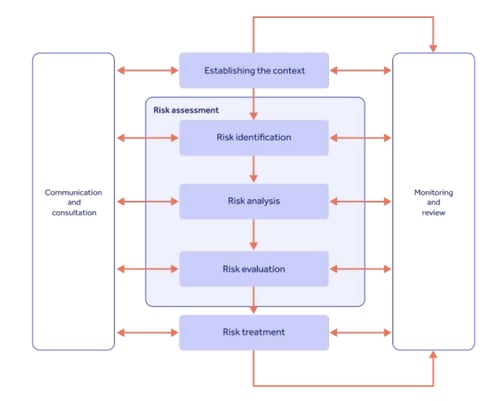
Since an optimized protocol is the key to reliable data and patient protection, ICH E6 R2 made it clear that a risk-based quality approach is vital to get repeatable GCP in place.
We'll go deeper into ICH E6 risk management below.
Download an NIDCR clinical quality management plan (CQMP) template
Updated roles & responsibilities
ICH E6 R2 didn't really tinker much with its original definition of the 'investigator' role. Investigators continued to have ultimate responsibility for trial execution.
The sponsor section, however, saw a considerable overhaul in 2016.
The new quality and risk focus noted above falls mainly on the sponsor's shoulders, while Section 5.2.2 of ICH E6 R2 prescribes extra oversight responsibilities for sponsors outsourcing their clinical activities.
Which brings us to...
Study monitoring and oversight
Sponsors take on 'ultimate responsibility' for trial data quality and integrity, even if they aren't directly involved in some trial functions.
The ICH GCP E6 (R2) guideline made it clear that sponsors require robust and documented oversight capabilities to keep an eye on their sponsored studies.
That doesn't just mean checking up on your CRO with an audit now and again. Even trial operations subcontracted by your CRO to another third party need to be considered.
FURTHER READING:
That means being able to plan, describe and document all trial data flows and their connected parties, as well as how trial activities will be measured and assessed.
Changes to study monitoring expectations constituted the biggest update in ICH E6 R2 back in 2016.
The addendum defined three types of monitoring and gave sponsors the freedom and flexibility to choose the format most appropriate - as long as the reasoning is documented in your monitoring plan.
1) On-site monitoring (the classic clinical trial format)
2) Centralized, 'off-site' monitoring with real-time data collection and review
3) A combination of the two
In return, the sponsor should receive frequent monitoring reports from their investigators, allowing them to take corrective and preventive action where necessary to address data gaps, trends, errors and other non-conformances.
Clinical trial communication & documentation
ICH E6 R2 placed more emphasis on sponsor-investigator communication, as touched on above.
The addendum also incorporated ALCOAC (but not the full wheelhouse of ALCOA+) principles in its 'Records & Reports' section, unlike the original ICH E6.
Trial documents and data should be attributable, legible, contemporaneous, original, accurate and complete. It's still worth embracing the entirety of ALCOA+ as you build your trial document management processes:
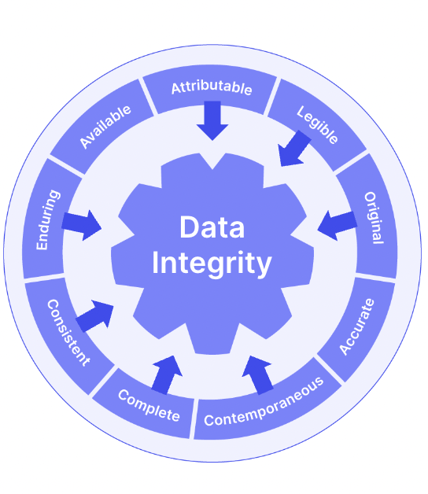
We've seen already that the risk-based quality focus of ICH E6 R2 has the ultimate goal of maximizing both subject wellbeing and data accuracy.
Airtight document control was therefore much more emphasized in R2, and both sponsors and investigators were expected to keep separate, but equally well-managed, document sets that are easily retrievable.
Even though investigators have to report trial data to their sponsors, as we've just seen, they retain overall control of and access to that data for the duration of the trial.
Essential clinical trial documents include:
- Case report form (CRF) data
- Investigator of Record (IoR) form or Form 1572
- CVs for PI and Sub-Investigators
- Licenses (as appropriate)
- Training records for all study personnel
- Protocol/amendment signature page
- IRB membership list or roster
- IRB approvals of protocol, consents, ads, handouts, etc.
- Communications between IRBs, sponsors, CROs as required
Technology and data management updates
Lots changed between 1996 and 2016, including the rapid digitization of life science processes.
Digital health technology (DHT), Clinical Trial Management Systems (eCTMS), Electronic Data Capture (EDC) systems and electronic quality management systems (eQMS) all now have critical roles to play in modern clinical trials.
Read the FDA's draft guidance on digital health technology for remote clinical data capture
ICH E6 R2 took that into account, recognizing the new use of electronic systems for managing trial data.
Risk-based validation of computerized systems was mandated, along with SOPs for their usage and application - from validation and setup to training, backups, indexability and retrieval, recovery, testing and so on.
And, as we've already explored, the sponsor took responsibility for the integrity of electronic records and data throughout the trial process - even if it sits in a CRO's system.

Trial sponsors should:
- Be aware of their territory-specific data integrity requirements, like 21 CFR Part 11 or EU Annex 11, since responsibility for regulatory adherence ultimately falls to them
- Take steps to strengthen their clinical trial approach where possible with a digital system, underpinned by automated functionality for key areas like e-signatures
- Scope the data processes of potential CROs (if outsourcing) and consider them when qualifying candidates
- Integrate data management principles like ALCOA+ into their trial activities as early as possible
CROs should:
- Consider explicitly designating Part 11 compliance as part of the transfer of sponsor responsibility to remove any ambiguity
- Optimize data management processes and embed ALCOA+ to become as attractive and trusted as possible to new sponsors
- Factor Part 11 and/or Annex 11 compliance into scoping decisions when looking for a new eCTMS, EDC or eQMS
Key changes from ICH E6 R2 to ICH E6 R3
What about the nine years between R2 and R3?
Like any regulatory revision, ICH E6 (R3)'s purpose is to modernize, harmonize and adapt currently accepted best practice.
As ICH put it during the build of R3:
"The development of E6(R3) will address the complexities of clinical trials in the current global regulatory climate..."
- ICH E6 (R3) Final Concept Paper
ICH didn't only want E6 (R3) to more closely align with new standards like E8 (R1), "Revision of General Considerations for Clinical Studies".
They also pointed to a 'perceived problem' of E6 (R2) failing to keep pace with, and fully satisfy the regulatory demands of, the latest clinical trial innovations.
Key changes since 2016 include:
- New trial designs
- Further technological advancement
- Breadth and depth of data capture and sources
- New types of testing facilities
- Proliferation of outsourced service providers and more complex clinical supply chains
E6 (R3)'s purpose is to respond with new content around:
- Electronic systems, data, documents and integrity
- Interventional and 'non-traditional interventional' trial formats
- The underlying principles of GCP
- Risk- and quality-based considerations for the protection of patient safety and data integrity
- 'Decentralized' and 'pragmatic' clinical trial elements
ICH E6 (R3), in its finished form, comprises 3 sections:
1) Overarching 'principles' document
2) Annex 1: considerations for interventional trials
3) Annex 2: further considerations for 'non-traditional interventional' trials
It's important to note, though, that R3 tweaks and tinkers with R2's content rather than representing a dramatic overhaul. For clinical teams making the transition, that's good news.
The first change from ICH E6 (R2) to ICH E6 (R3) can be gleaned from the principles listed above.
Principle 6, about building quality into clinical trials with a quality-by-design approach, takes the quality focus of R2 and builds on it.
Where R2 wanted clinical teams to 'ensure the quality of every aspect of the trial' with an assurance approach, R3 pushes for a more proactive, forward-thinking and considered approach to quality, where it's baked into the very fabric of the trial design and operation.
That means training procedures, protocols, SOPs and your wider document stack should only be constructed after a clear quality-based focus has been established.
What does this mean? The other key change to R3 - deeper focus on risk, seen in Principle 7 - helps illustrate what's required.
Any critical risks and quality factors which could threaten either of those two main GCP goals of patient safety and data integrity should be pinpointed as your very first set of work.
This proactivity, in turn, should inform a strategy of clinical design built on measurable quality characteristics and risk metrics, underpinned by multivariate analysis and clear lines of communication.
And your risk-based thinking should drive proportionality, with maximum planning effort focused on the processes and data streams with the greatest impact on patient safety and the outcome of the trial.
The final outcome of this quality and risk approach?
E6 (R3) moves beyond the ALCOAC data integrity requirements of R2 and focuses on the broader output requirement of data reliability - results should be able to be accepted with 'confidence', and in turn 'support good decision-making'.
A beneficial side-effect of this lean, proportional risk-based focus is the acceleration of evidence generation, and ICH hopes the new emphases of R3 will help steer IPs through efficient, optimized trials faster than ever.
Download our clinical trial quality by design (QbD) guide to start getting ready
Risk-based approach in ICH E6 R3
Risk, then, is at the heart of ICH E6 R3's good clinical practice framework, with patient and data risk management at the forefront.
And studies show why a proactive, risk-based approach is so crucial. A 2013 PwC study found that taking the time to properly map out and address clinical risks adds an extra 2% to your initial costs, but in return:

Risk-based thinking should cascade into all of your clinical risk management activity, beginning with assessment and planning.
Risk assessment and planning
Start early. Critical data and processes, and connected risks that jeopardize patient safety and data integrity, should be pinpointed as early as possible - even if your protocol is still in draft, for instance.
The protocol itself can then be molded and tweaked to address any inherent risks, allowing quality by design to be baked into the operation while it's still in flux, and before other bodies like a CRO get involved downstream.
ICH E6 R3 pushes for collaborative, cross-departmental and cross-company risk planning, too.
Sponsors, CROs and even - as touched on above - computerized system vendors all have their role to play in actively assessing, documenting and planning against risks.
It's wise to appoint a dedicated risk manager at this stage.
And remember: you'll encounter different risks at different stages of your clinical trial. Risk assessments should therefore be targeted, stage-specific and frequent, with particular focus on key transition moments like moving from one clinical phase to another.
Risk identification & analysis
Now we have our risks, it's time to analyze their potential impact.
ICH E6 R2 offered some help here by introducing so-called 'quality tolerance limits' (QTLs): a handful of 'sweet spot' topline metrics to measure throughout your clinical trial and to assess your risks in relation to. These remain relevant in ICH E6 R3.
QTLs are used 'to identify systematic issues that can impact subject safety or reliability of trial results'.
Classic risk analysis tools and techniques you can apply at this stage are:
- FMEA
- FMECA
- FTA
- HAZOP
- PHA
- Ranking and filtering
- Control charts
- Pareto charts
- Process capability analysis
And so on.
Risk evaluation & control
The assessed severity of a risk's impact should now be combined with its likelihood and detectability to evaluate the controls that'll be required to reduce that risk to an acceptable level.
Obviously, the more likely and severe the risk, the more controls required to prevent it actualizing.
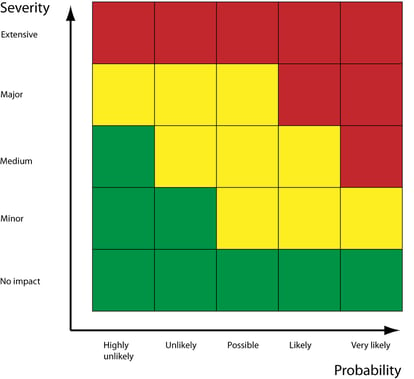
Your risk controls, such as corrective and preventive actions (CAPAs), should then be applied to either mitigate, avoid or entirely eliminate the risk.
Your residual risk score (i.e. the likelihood and severity of the risk after controls are added) should be assessed and monitored throughout the reduction process.
And, of course, key risk indicators (KRIs) need to be fed into your clinical oversight planning to allow you to keep an eye on them.
Which leads us nicely to...
Risk review & monitoring
ICH E6 R2 encouraged constant review and monitoring of risk actions to ensure risks remain acceptable and adequately treated, and we've seen that proportionality and proactivity were emphasized in ICH E6 R3.
Your risk management should be an iterative and ongoing process throughout the entire lifecycle of your clinical trial - so give yourself the ability and cadence to revisit and update your risk management plan as new insights emerge, risk scores alter and protocols evolve.
Clearly documented and communicated risks should be shared between sponsor and investigator, and vice versa, to allow collaborative safeguards to be established as required as the trial unfolds.
Implementation of ICH E6 R3
Now we've touched on the broad ingredients and risk requirements of ICH GCP E6 (R3) guideline, how do we get it baked into our clinical processes for repeatable GCP?
ICH E6 R3 maps out 11 principles of good clinical practice (down from 13 in R2) which can be a helpful starting point as you look to embed compliance.
Strategies for implementing ICH E6 R3
Think of the 11 GCP principles as your topline strategic objectives for E6 compliance. They are:
1. Clinical trials should be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with GCP and applicable regulatory requirement(s).
Clinical trials should be designed and conducted in ways that ensure the rights, safety and wellbeing of participants.
2. Informed consent is an integral feature of the ethical conduct of a trial.
Clinical trial participation should be voluntary and based on a consent process that ensures participants (or their legally acceptable representatives, where applicable) are well-informed.
3. Clinical trials should be subject to an independent review by an institutional review board/independent ethics committee (IRB/IEC).
4. Clinical trials should be scientifically sound for their intended purpose and based on robust and current scientific knowledge and approaches.
5. Clinical trials should be designed and conducted by qualified individuals.
6. Quality should be built into the scientific and operational design and conduct of clinical trials.
7. Clinical trial processes, measures and approaches should be implemented in a way that is proportionate to the risks to participants and to the importance of the data collected.
8. Clinical trials should be described in a clear, concise and operationally feasible protocol.
9. Clinical trials should generate reliable results.
10. Roles and responsibilities in clinical trials should be clear and documented appropriately.
11. Investigational products used in a clinical trial should be manufactured in accordance with applicable Good Manufacturing Practice (GMP) standards and be stored, shipped, handled and disposed of in accordance with the product specifications and the trial protocol.
These broad coordinates of GCP should cascade down into your E6 compliance strategy in a kind of 'golden thread' approach.
Some key ingredients you'll need to get in place to achieve the principles are:
- Monitoring plan
- Robust adverse event and safety reporting
- Clear documentation and communication SOPs and structure
- Data collection and reporting procedures
- Risk management plan
- Clear protocol with mapped roles and responsibilities for sponsor and investigator
- Quality management system
- Complete compliance with the protocol, deviating only for admin/
logistical changes or where there is an immediate danger posed to subjects, and notifying the IRB, sponsor and (if necessary) the regulatory authorities of deviations - Control of investigational product usage and storage
- Randomization and unblinding
- Informed consent process
- Data integrity and ALCOAC (ideally ALCOA+)
A 2015 Pfizer case study is a good example of how to map those broad GCP principles into actionable, tactical clinical ingredients and KPIs, or 'outcome components':
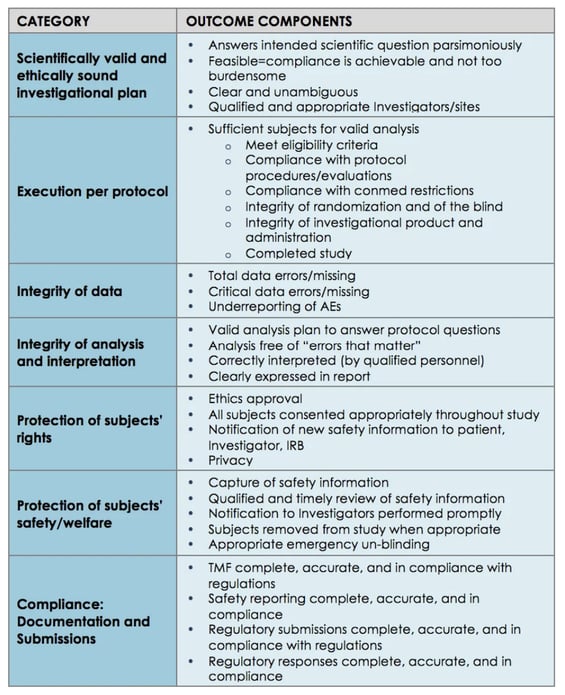
Challenges & solutions
The most common challenges threatening GCP, and standing in the way of your ICH E6 R3 compliance, are:
Building a risk-based approach
Pivoting your entire culture towards constant risk focus isn't easy and demands clear communication and a connecting system uniting sponsor and investigator as much as possible.
Manual document management
Reliance on paper, time-heavy control and collation of documentation, no verification of data as it’s recorded, weak audit trailing.
Poor documentation
The structure of documents themselves may be substandard, providing an insufficient level of guidance or detail and encouraging written procedures to be ignored or worked around.
Poor training
Protocol and GCP training isn’t properly planned, executed or recorded. Standardized and competence-based training is performed manually and struggles to keep up as staff are rotated in and out of trials.
Fuzzy protocols
A clear, documented sponsor-investigator relationship is crucial. Unsuccessful trials are often bogged down by unclear roles and responsibilities.
No listening
Inspectors and patients are often an untapped goldmine of feedback and guidance. When inspector reports and patient advocacy groups aren’t harnessed, mid-trial corrections and improvements become more difficult to make.
Weak QMS
A disconnected or heavily manual QMS with limited investigational involvement and oversight, and inappropriate SOPs, makes a harmonized, robust quality approach much more difficult to instil. Quality issues remain undetected and can grow into a trial-ending event further down the line.
Low integration of quality and ops
Clinical trial quality focuses on performance, audits, deviation control and, of course, management of quality through QC and QA activity.
Clinical trial operations focus on the nuts and bolts of the trial experience: staffing, data, protocol execution and scientific review. Where these two streams run in parallel without touching, robust quality by design and ICH E6 compliance isn't optimized.
As a solution to all these problems, consider investing in a cloud-based electronic quality management system (eQMS) as early as possible in your clinical journey.
eQMS platforms unite document, training, process, supplier and analytic management in a single, remotely accessible source of truth.
Sponsors can share access with CROs and vice versa, as well as benefiting from automatic data integrity guardrails and full traceability of key quality metrics like training, document and CAPA statuses or KPIs, KRIs and QTLs.
And the results of ending time-consuming admin, and freeing up time for proactive quality improvement, can be dramatic...
See how a clinical trial provider in Dublin unlocked unprecedented quality maturity
Best practices for compliance with ICH E6 R3
Like any regulatory guideline, ICH E6 R3 compliance can be simplified and accelerated by embedding best practice.
And getting things right the first time around is important: it costs about $140,000 to amend a protocol once your trial gets to Stage II.
Let's explore some elements that you'll need to think about.
Ensuring quality by design
We've seen how E6 R2 and E6 R3, unlike the original E6, put quality and risk front and center of your clinical planning.
QbD isn't explicitly required by the guideline, but it makes things a whole lot easier.
Baking quality into your protocol as early as possible slices the risk of substantial amendments and errors further down the line.
That means:
- Planning for the frequent review, measuring and communication of quality and risk 'signals' like QTLs, KPIs and KRIs
- Getting mechanisms in place for CAPA and change management
- Securing active participation from senior management, including overview and allocation of resources
- Ensuring all elements of the trial, from sponsor to CRO and trial site, are constantly audit-ready and GCP-compliant
Look to ICH Q10, the pharmaceutical quality standard, for some inspiration and cross-fertilization around the concepts of quality by design.
Selecting and managing vendors and partners
We've seen how ICH E6 R3 makes risk management a shared, proactive, multi-party commitment across the clinical lifecycle.
Who you choose to work with is therefore vitally important.
A robust supplier management strategy should be in place, allowing partners like CROs to be carefully vetted, onboarded, then held accountable for key investigational activities.
Remember that a vendor's risk tolerance levels and risk management approach may be different from yours, so ensure there's no dissonance before you start working with them.
Also remember that not all clinical sites have their own quality program in place - only the largest ones have independent programs, with the rest receiving quality standards and metrics on a case-by-case basis from sponsors and CROs. Clear communication of expectations is therefore vital.
Key vendor management ingredients to get in place are:
- Onboarding and assessment processes
- Shared quality agreements (QAgs) for sponsors, CROs and sites
- Audit cadences and the 'right to audit'
- Quality management plan and agreed quality metrics within site agreements
Utilizing technology in clinical trials
We saw how ICH E6 R2, with its emphasis on deeper monitoring and electronic data management, emerged in response to the more digital clinical environments of the 2010s. ICH E6 R3 built on that by mapping out fresh requirements for electronic systems and documents.
That doesn't mean the latest clinical technology is now mandatory for you to operate, but it logically follows that the industry is now more expectant and encouraging of investment in this technology than it was in 1996.
(More on that below.)
And why not? Modern technology accelerates the clinical experience while strengthening and systematizing quality - so the right investment can lift your operation to the next level and provide a better experience for your subjects and your operating partners.
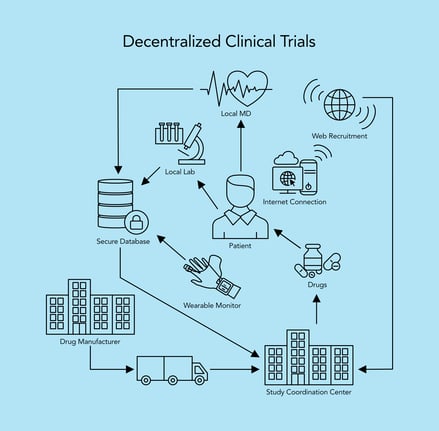 The modern 'decentralized' clinical trial (Source: HTEC Group)
The modern 'decentralized' clinical trial (Source: HTEC Group)
Here are some key aspects of modern clinical trial technology you should consider to augment your ICH E6 compliance:
-
Electronic Data Capture (EDC)
EDC systems have replaced traditional paper-based data collection methods in clinical trials. These systems enable electronic capture, management and storage of clinical trial data, allowing for more efficient data entry, real-time data monitoring and improved data quality
-
Electronic Patient-Reported Outcomes (ePRO)
ePRO tools enable patients to report their symptoms, treatment experiences and quality of life directly through electronic devices like smartphones or tablets. This method eliminates the need for paper diaries and provides more accurate and timely patient-reported data
-
Electronic quality management systems (eQMS)
eQMS platforms make it easier to meet the quality and risk management demands of ICH E6 R3 by automating and centralizing key activity like analytics, documents, training and suppliers in the cloud. An eQMS takes care of quality admin for you, freeing up time for more proactive, improvement-based quality work
-
Wearable devices and sensors
Wearable devices such as fitness trackers, smartwatches and biosensors are increasingly used in clinical trials to collect continuous physiological data from participants. These devices can monitor vital signs, activity levels, sleep patterns and other relevant health parameters, providing investigators with objective real-time data
Learn how a wearable manufacturer hit new quality management heights
-
Telemedicine and remote monitoring
Telemedicine platforms and remote monitoring technologies allow for virtual visits, remote patient monitoring and teleconferencing between investigators and participants. These tools became particularly valuable during COVID-19, enabling the continuation of clinical trials while ensuring patient safety and reducing the need for in-person visits
-
Data analytics and Artificial Intelligence (AI)
Advanced data analytics and AI techniques are being employed to process and analyze the vast amount of clinical trial data generated. AI algorithms can help identify patterns, predict outcomes and uncover valuable insights from complex datasets, ultimately improving decision-making and enhancing trial efficiency
LISTEN TO OUR PODCAST:
Hear the CEO of NovaDiscovery talk about 'in silico' AI-powered trials!
-
Patient recruitment & engagement tools
Technology has also revolutionized subject recruitment and engagement for clinical trials. Online platforms, social media and mobile apps can now be utilized to reach a broader pool of potential participants, provide trial information and improve communication and interaction with study subjects
-
Blockchain
Blockchain technology is being explored for enhancing data security, integrity and privacy in clinical trials. By leveraging distributed ledger systems, blockchain can ensure transparency, immutability and traceability of clinical trial data, reducing the risk of fraud and boosting subject confidentiality and participation
-
'Adaptive' trials
Modern clinical trial technology supports adaptive trial designs, where study protocols can be modified in response to accumulating data during the trial. This flexibility allows for efficient allocation of resources, faster identification of effective treatments, and early termination of ineffective interventions
Continuous monitoring and performance metrics
Above all else, ICH E6 R3 puts its foot down on 'fire-and-forget' clinical trials.
A handful of QTLs, KPIs and KRIs should be established, then proactively focused on across the clinical lifecycle with collaborative input from both sponsor and investigator.
In turn, this should drive agile quality-focused management of your clinical trial with an 'adaptive' approach (noted above) employed to continually tweak and refine your trial as it develops.
Future of clinical trials and ICH E6 R3
It would be foolish to assume that ICH E6 R3, when it goes live, will be the final blueprint for 21st-century clinical trial best practice.
As trial technology, formats and objectives evolve, we can fully expect an R4 or R5 further down the line.
Since trials can stretch for years, this can place clinical actors in an ever-moving regulatory environment - so keeping abreast of industry developments and expectations is key!
