The 3 FDA medical device classes: differences and examples explained
There are distinct FDA medical device classes for a reason. If you're getting a replacement heart valve, you probably want to know the device going into your chest went through a more rigorous approval process than a pair of surgical tweezers. Thankfully, the FDA agrees with you.
As you plan a route to market for your medical device, it's important you understand early what each class means, which class your device falls into, and what your regulatory pathway looks like as a result.
We've broken down all of the medical device classes in detail to help you understand them all.
Table of Contents
- What are FDA medical device classes?
- Medical devices examples
- Differences between FDA medical device classes
- What is an FDA Class 1 medical device?
- What is an FDA Class 2 medical device?
- What is an FDA Class 3 medical device?
- How to classify medical devices
- Accelerate your route to market with the right tools
What are FDA medical device classes?
The FDA regulates all medical devices marketed in the U.S. and assigns every single one a 'class' based on its risk profile: that is, the potential threat posed to the safety and health of a patient were something to go wrong with it.
These classes are of vital importance to quality and compliance professionals: they set the regulatory structure and pathway your device must follow to secure market access.
FDA medical device classification
How many medical device classes are there?
Any medical device approved by the FDA Center for Devices and Radiological Health is classified into one of three classes: either Class 1, 2 or 3, depending on its risk, invasiveness and potential impact on patient health. These classes are also written as Roman numerals for official FDA usage: Class I, II and III.
Class 1 represents devices with the lowest risk profile, while Class 3 devices pose the highest risk.
But where are the lines drawn between each of these three classes, and why?
There's an enormous difference in the optimal paths to market, depending on how your device is grouped. Class 1 devices, obviously and logically, are subject to far fewer regulatory requirements than Class 2 or 3 devices.
Medical device definition
Before we move on, it's useful to backtrack and ensure we're familiar with what a medical device actually is.
What is a medical device?
The lines aren't always clear-cut between what is and isn't: scalpels and heart monitors are obviously medical devices, but you might be surprised to learn that toothbrushes also fall into the medical device category.
In the 21st century, there's also hardware vs software medical device breakdown to consider. Medical devices no longer have to be purely physical, operating room applications - they can live entirely on servers and computer screens.
And medical software is sometimes mistakenly identified as a medical device, when that categorization heavily depends on the type and application of the software.
Here's a handy guide to help you understand the medical device definition, and what a medical device is, in more detail:

Medical devices examples
To bring this distinction to life, let's take a quick look at the types of medical devices that fall into each risk category.

These medical device examples help bring the class hierarchy into focus.
We can see that the low-risk devices with minimal patient impact, like bandages and toothbrushes, fall into Class 1 on our medical device list.
Examples of medical devices with a high-risk category include highly invasive and life-critical applications like ventilators, while medium-risk devices like syringes and insulin pumps fall into the middle category of Class II.
It's clear, then, that risk sits at the heart of FDA medical device classes and how they're structured.
Types of medical devices
The type of medical device determines whether it falls into Class 1, 2 or 3.
How critical a device is to a particular process is an important way of calculating risk and giving it a type.
This is known as its 'intended use': does the device directly treat a symptom, or just guide a doctor towards helping them treat it?
The significance of that process to the patient, and how the device's 'indications for use' will make an impact on that patient's wellbeing, matter too for determining risk. Is the device playing a direct role in fixing a patient's eyesight, for instance, or in saving their life after being hit by a car?
These medical device example groups are made clearer when we look at how SaMD medical devices fall into Class 1, 2, 3 or 4 groups in the International Medical Device Regulators Forum (IMDRF)'s framework.
You can see how the importance of the healthcare condition interacts with the device's contribution to produce an appropriate risk score.

Differences between FDA medical device classes
Before we get into classes and their differences, we need to understand specialties.
The FDA has classified over 1,700 distinct types of medical devices, organized in the Code of Federal Regulations (CFR) according to 16 grouping 'specialties'. Classifying your device according to one of these specialties is the first step to understanding whether you are manufacturing a Class 1, 2 or 3 medical device.
The 16 specialties are:
- Anesthesiology
- Cardiovascular
- Chemistry
- Dental
- Ear, nose and throat
- Gastroenterology and urology
- General and plastic surgery
- General hospital
- Hematology
- Immunology
- Microbiology
- Neurology
- Obstetrical and gynecological
- Ophthalmic
- Orthopedic
- Pathology
- Physical medicine
- Radiology
- Toxicology
After finding your specialty, it's time to move into classes.
As we'll see, the class you sit in determines your regulatory pathway and the level of oversight the FDA requires.
For example, Class 1 devices, and a handful of exempt Class 2 devices, are exempt from premarket notification and approval processes, while Class 3 and the majority of Class 2 devices require active dialog with and investigation by the FDA.
Another type of device may be exempt from some regulatory controls, depending on its intended use. A humanitarian device exemption may be issued for devices intended to help with rare diseases, as the sample size is often too small to obtain enough clinical evidence to meet the FDA’s reasonable assurance standards for 'safety and effectiveness' laid out in Sections 514 and 515 of the Federal Food, Drug and Cosmetic Act.
However, all devices regulated by the FDA are subject to current Good Manufacturing Practice (cGMP) requirements for registration, labeling and quality.
So, with those caveats in mind, how do you know if your device is Class 1, 2 or 3, and whether or not you’re required to undergo premarket notification?
Let's take a look at each class in turn.
What is an FDA Class 1 medical device?
The FDA defines Class I devices as:
"not intended for use in supporting or sustaining life or of substantial importance in preventing impairment to human health, and they may not present a potential unreasonable risk of illness or injury."
These devices are the most common class of devices regulated by the FDA, constituting 47% of approved devices on the market.
Class 1 devices have minimal contact with patients and low impact on a patient's overall health. In general, Class 1 devices do not come into contact with a patient's internal organs, central nervous system or cardiovascular system.
These devices are subject to the fewest regulatory requirements.
Class 1 medical device examples
- Electric toothbrush
- Tongue depressor
- Oxygen mask
- Reusable surgical scalpel
- Bandages
- Hospital beds
- Non-electric wheelchair
Bringing Class 1 medical devices to market
As you might expect, Class 1 devices are the fastest and easiest to bring to market since they present the lowest amount of risk to the patient and are rarely critical to life-sustaining care. The majority of Class I medical devices are exempt from FDA requirements for Premarket Notification (510(k)) and Premarket Approval (PMA).
Class 1 devices are not exempt from FDA General Controls, a series of commands which applies to Class 1, 2 and 3 medical devices. The provisions of General Controls address adulteration, misbranding, device registration, records and good manufacturing practices.
Medical device manufacturers who fall into Class 1 are still required to implement a quality management system and follow standards to ensure a quality product.
What is an FDA Class 2 medical device?
Class 2 medical devices are more complicated than Class 1 devices and present a higher category of risk because they are more likely to come into sustained contact with a patient. This can include devices which come into contact with a patient's cardiovascular system or internal organs, and diagnostic tools.
The FDA defines Class II medical devices as:
“devices for which general controls are insufficient to provide reasonable assurance of the safety and effectiveness of the device.”
Class 2 medical device examples
- Catheters
- Blood pressure cuffs
- Pregnancy test kits
- Syringes
- Blood transfusion kits
- Contact lenses
- Surgical gloves
- Absorbable sutures
Bringing Class 2 medical devices to market
Controls vary depending on the device, but according to the FDA, can include:
- Device performance
- Post-market surveillance
- Patient registries
- Special labeling requirements
- Premarket data requirements
- Guidelines
Class II medical devices are subject to the same General Controls mentioned above, as well as Special Controls. These regulations depend on the device and may include special labeling requirements, patient registries and performance standards.
RELATED READING: 4 major Class 2 medical device requirements
Most importantly, most Class 2 devices come to market using the premarket notification 510(k) process. The 510(k) is a complex application to the FDA, which demonstrates that a device is safe and effective by demonstrating that the device is equivalent to another device which is on the market.
This process involves showing 'substantial equivalence' to the existing marketed device, known in FDA parlance as the 'predicate'. This doesn't mean the devices need to be identical, but they require significant similarities in use, design, materials, labeling, standards and other characteristics.
The FDA released an exemption list in early 2018 which exempts over 800 device types from the 510(k) process. If you have a generic Class 2 medical device, you can discover whether it is exempt from a 510(k) filing by reading our guide.
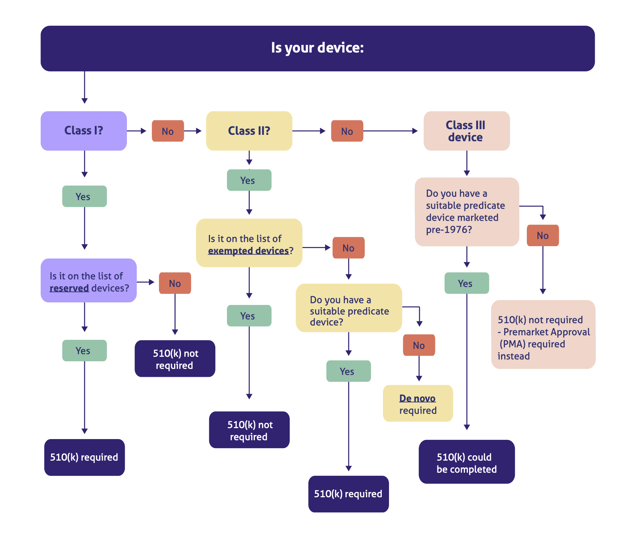
Because a 510(k) could theoretically be completed for any device class within certain parameters, we built this handy decision tree map to help you determine if you need one:
What is an FDA Class 3 medical device?
The FDA defines Class 3 devices as products which:
“usually sustain or support life, are implanted or present a potential unreasonable risk of illness or injury."
Just 10% of the devices regulated by the US FDA fall into Class III. This classification is generally extended to permanent implants, smart medical devices and life support systems.
Class 3 medical device examples
- Breast implants
- Pacemakers
- Defibrillators
- High-frequency ventilators
- Cochlear implants
- Fetal blood sampling monitors
- Implanted prosthetics
Bringing Class 3 medical devices to market
Class 3 devices are subject to all General Controls and the FDA's Premarket Approval (PMA) process. The FDA writes that 'general and special controls alone are insufficient to assure the safety and effectiveness of Class 3 devices'.
Appropriately, the PMA is the most intensive type of device marketing application required by the FDA.
As you can see in our decision tree above, some FDA Class 3 devices may qualify for the 510(k) route if you can find a suitable predicate marketed before the Medical Device Amendments of 1976. But as time goes by, this pathway is getting more and more unlikely and rare, since the FDA frowns on the use of predicates any older than a decade. The PMA is therefore by far your most likely option for a Class III medical device.
The PMA process and premarket review require a rigorous study of your Class 3 medical device to prove safety and effectiveness through the development of a data-driven benefit/risk profile.
The PMA process generally involves clinical trials, and significant time and resources for sufficient data collection. The FDA will also perform a substantive review of your quality system during this time.
RELATED READING: The difference between premarket notification 510(k) and premarket approval
How to classify medical devices
The first step towards classifying your medical device is to navigate the FDA classification regulations, the list of 16 categories for medical devices according to medical specialization.
As an example, we'll show you the steps to identify the classification of a blood pressure alarm. The device is classified under Category 870: cardiovascular devices.
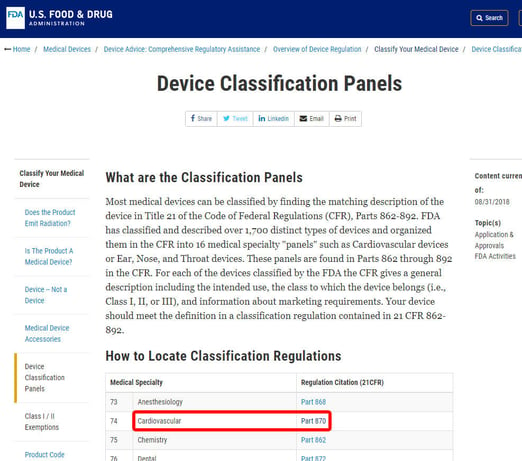
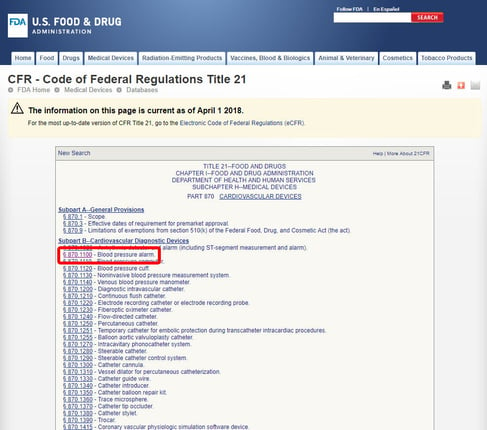
Once you've located the relevant medical specialty, click on the category, and navigate the list of devices until you find an equivalent and the associated device code.
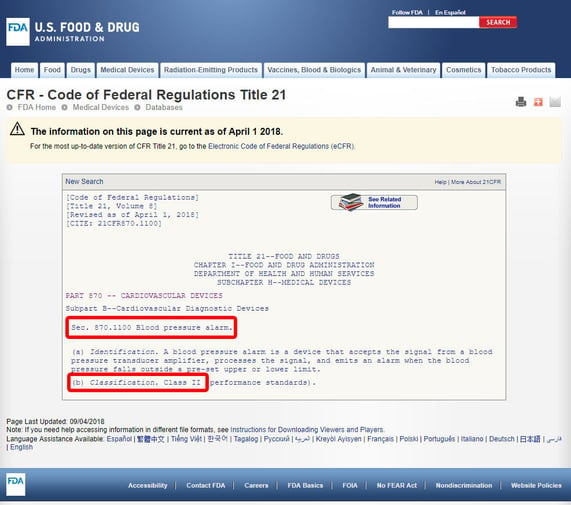
Click on the device code and open the guidelines. The device classification is listed under section (b). In this case, we can see that we're dealing with a Class 2 medium-risk device.
If your device lacks a listed equivalent among the 1,700 devices classified by the FDA, it is most likely an innovative device without a substantial equivalent, putting you in Class 3 medical device territory.
Accelerate your route to market with the right tools
Complex FDA clearance processes like the 510(k) or premarket approval demand vast amounts of device data and demonstrable proof of an airtight, high-performing quality management system.
Legacy quality management tools like paper and spreadsheets clutter, complicate and slow down your premarket processes and make clearance more difficult.
Whatever your medical device class, consider investing in a dedicated electronic quality management system (eQMS). Modern eQMS tools like Qualio centralize your Class 1, Class 2 and Class 3 medical device data and automate key quality processes, accelerating FDA compliance readiness by months.
Book a demo to see how it works!
