PMA meaning: understanding FDA pre-market approval
The FDA PMA process is a crucial regulatory gateway for companies bringing high-risk medical devices to the US market.
But "what is a PMA?" is a frequently-asked question. The PMA process is complex, stringent and often misunderstood.
This blog post maps out everything you need to know about securing FDA PMA approval for your organization.
Table of Contents
What is PMA?
The FDA PMA process is a US regulatory pathway for the approval of high-risk medical devices before they can be marketed and sold.
The PMA process is applicable to medical devices that are considered to have a high risk to patients, such as life-sustaining or life-supporting devices.
The FDA PMA process involves a comprehensive review of scientific data and evidence submitted by the device manufacturer.
You need to provide detailed information about your device's design, manufacturing process and performance and clinical data, demonstrating safety and efficacy throughout. The FDA then assesses this information to determine whether your device meets the required standards for clearance.
The PMA process, reflecting the high-risk nature of the devices it scrutinizes, is rigorous and typically involves multiple stages of review, including a quality management system review as well as a thorough scientific and clinical evaluation, usually including clinical trials.
It also includes inspection of your manufacturing facilities. The FDA assesses the benefits and risks of your device based on the data provided, as well as the potential impact on patient health and safety.
Upon successful completion of the PMA process, the FDA clears your device for marketization, allowing it to be sold and used in the United States.
PMA definition
'PMA' simply stands for 'pre-market approval'.
The logic is clear: the FDA requires pre-market approval for high-risk medical devices to ensure their safety and effectiveness before they enter the bodies of patients.
No PMA? No legal ability to market your high-risk device.
PMA meaning
Completing a PMA means that your medical device must pass through a comprehensive safety review process.
FDA PMA approval proves that the FDA has determined your device to be safe and effective for its intended use and, although high-risk by nature, offers sufficient patient benefit alongside its controlled risk to pose a net benefit to society.
FDA medical devices classification
The FDA categorizes all medical devices marketed in the US into three broad risk categories:
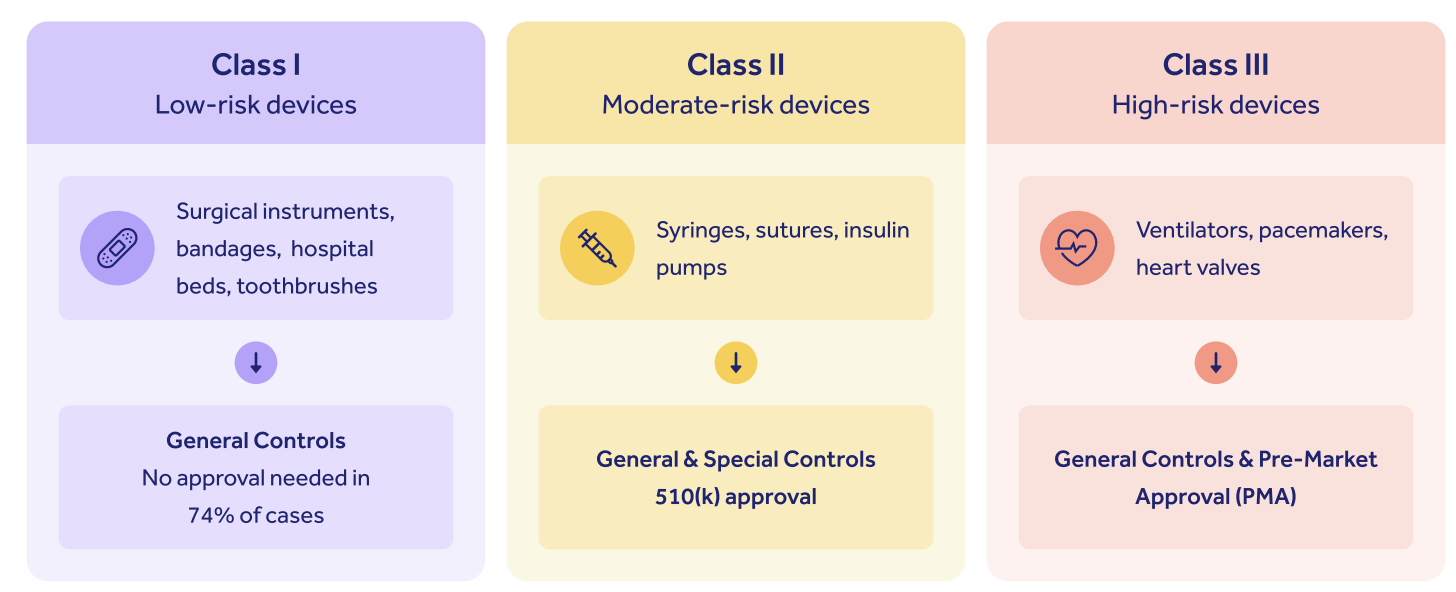
As you can see, while low-risk Class I devices require only general medical device controls with no regulatory oversight, and Class II devices require Special Controls alongside a 510(k) submission, it's Class III high-risk devices that fall under the FDA PMA bracket.
Class III devices constitute around 10% of all medical devices marketed in the US.
This tripartite regulatory structure embodies the FDA's emphasis on appropriate, targeted and risk-based oversight of life science products.
The higher your risk class, the higher the burden of proof that falls on you, the manufacturer, to prove to the FDA that your device is safe and effective.
The FDA PMA approval is therefore the most difficult and complex of all medical device approval processes in the US.
FURTHER READING: The 3 FDA medical device classes: differences and examples explained
Do Class II devices need PMA?
No, Class II medical devices in the US do not require completion of the FDA PMA process.
Let's look in more detail. This handy decision flowchart should help you determine your regulatory pathway:
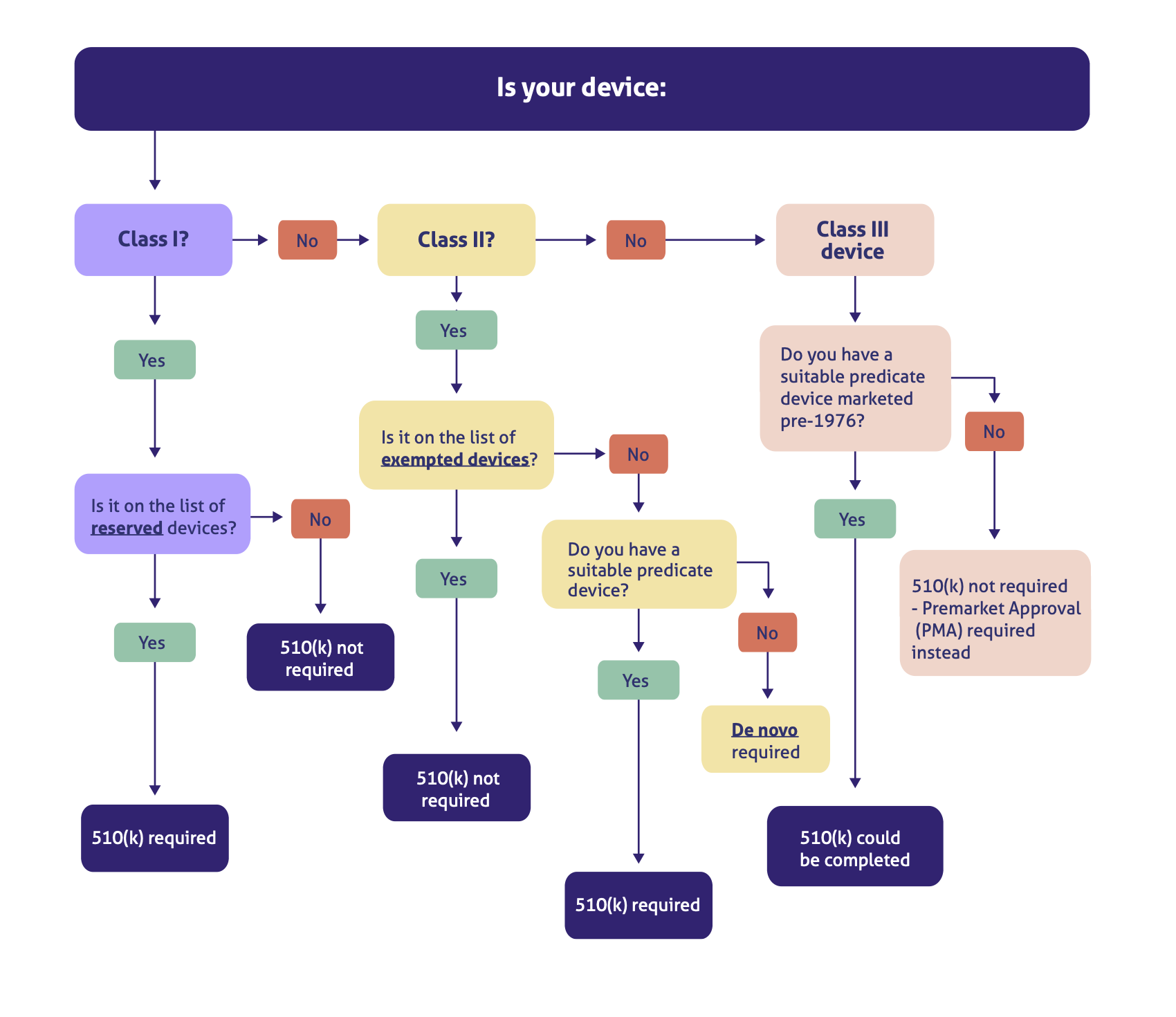
As you can see, while there are some exceptions here to our general three-part rule, such as some Class I devices needing a 510(k) and some innovative Class II devices needing a de novo, the alignment of 'PMA' and 'Class III' is clear.
Only Class III device manufacturers need to worry about the FDA PMA approval process.
The reason, as we've touched on above, is that any devices with a lower level of risk simply don't require the same extensive level of FDA oversight that a PMA approval demands.
So before you begin your FDA PMA preparations, make absolutely sure your device falls into the Class III category. (More on that below.)
Do all Class III devices require a PMA?
Yes, all modern Class III medical devices are required by the FDA to pass through the PMA process before marketization.
But here the plot does admittedly thicken a little, because there is a potential second option.
Because the 510(k) clearance process works by proving substantial equivalence to a medical device already on the market, it is possible to pass through the 510(k) process rather than the PMA process if you can find a high-risk device that's substantially equivalent to yours.
But the catch is that it must be a device marketed before the 1976 Medical Device Amendments to the FD&C Act.
Since high-risk Class III devices like pacemakers and heart valves are most subject to innovation and update, it's unlikely you'll be bringing a device to market that's substantially equivalent to mid-'70s technology.
And at any rate, the FDA frowns on the use of predicates any older than a decade or so.
This route therefore appears increasingly (and vanishingly) unlikely as a regulatory pathway option for Class III manufacturers.
It's therefore safe to generalize and say that all modern Class III devices will require a PMA approval completion.
What is an example of a PMA device?
Not sure if you need a PMA?
The FDA defines Class III medical devices as those that:
... usually sustain or support life, are implanted, or present potential unreasonable risk of illness or injury...
Class III medical devices that fall under the FDA PMA umbrella include:
- Active implantable medical devices (AIMDs)
- Artificial organs
- Pacemakers
- Defibrillators
- Ventilators
- Heart valves
- Breast implants
- Implanted prosthetics
Still not sure? Head to the FDA classification database and look for similar devices
Who can submit a PMA application?
Anyone in your business can submit an FDA PMA application, as long as your business has the rights to the device or has 'authorized access' to the data you'll need for the application.
Speaking of which, let's dive into what you'll need to show the FDA.
Required data for PMA approval
Needless to say, the rigor and stringent oversight needed for Class III devices translates into a lot of data requirements.
The topline requirements are as follows:
- Cover letter identifying your submission type
- Summary of major submission points (10-15 pages approx.)
- Device description
- Alternatives to your device for the condition/disease you're treating
- Marketing history
- Technical data summary and conclusions
- Detailed device description
- References to performance/voluntary standards applicable to any aspect of your device's safety and efficacy
- Technical data
- Non-clinical laboratory study results
- Clinical trial results
- Justification of a single investigator (if you've only used one)
- Any published reports about your device
- Samples if requested, or location where one can be examined
- Proposed labeling, including promotional materials if available
- Financial certification or disclosure statement
TOP TIP: If you're a small business, complete FDA Form 3602 (or 3602A if you aren't American) to register as one.
Your PMA application will then cost $110K rather than $441K!
The PMA application process explained
It's important to note the different types of FDA PMA applications, since these influence what exactly your submission process looks like.
It's most likely that you'll go down the traditional PMA route. This is for Class III devices that have already completed clinical tests, and therefore for companies that already have the requisite clinical data to give to the FDA.
But you can also start your PMA early, before clinical testing is complete, and go down the modular PMA route. This 'one-at-a-time' approach lets you complete sections of your PMA as your device's development and testing unfolds, submitting them individually into an overarching 'Shell'. Once all ingredients are submitted, you have a complete FDA PMA submission ready for review.
A third option is the Product Development Protocol. It follows a similar vein of substantial equivalence that we saw in the 510(k) submission process, and it's for companies bringing a device to market that uses technology already widely established and understood. A PDP maps out in advance the design and development milestones expected of you, which you can then work through at your own pace until completion.
The final, and rarest, option is the Humanitarian Device Exemption (HDE) process. It's for devices used to treat rare conditions (affecting <8000 Americans annually). The HDE process places limits on profitability, allowing it only if intended for use on children, and even then only up to a designated finite amount.
Whichever option you go for, ensure your data and scientific writing is robust, logical and easy to follow and trace.
The FDA expects an electronic 'eCopy' submission of your PMA, ensuring all the data ingredients above are ticked off and included. Once you've submitted, it's time to sit back for the review process.
The 4-step PMA review process
It can take around 6 months for your PMA review to complete once you've submitted everything to the FDA and it's been filed.
Your FDA PMA approval process will pass through 4 review stages.
1. Initial review
The FDA will begin their review of your application with an administrative once-over.
In short, does your eCopy contain all the necessary information for them to review, file and make an informed decision?
Common mistakes which can trigger a knockback at this early stage include:
- False or unsubstantiated statements
- Incomplete or missing application sections
- No justification for omitted sections
- Simultaneous 510(k) application for the same device
2. Substantive review
Assuming your submission is in order, it'll be filed by the FDA and the 180-day clock will start ticking.
You'll be informed by the FDA if they require any extra information to progress your FDA PMA approval at this point
You can also request a status update meeting if you wish. This should be requested no later than 70 days after filing, and will take place in the first 100 days.
3. Panel review
Not every device going through the FDA PMA approval process will need a panel review, but the FDA could set one up if your device is particularly new and innovative.
In this case, an advisory committee will review your application together in a public meeting.
4. Decision announcement
Now for the exciting part. Within 180 days of Phase 2 beginning, you'll be notified of the FDA's decision your PMA application.
You'll receive either:
- An approval order - well done!
Your device has come through with flying colors and is ready for marketization. You'll need to send the FDA your final labeling before you begin to market, but otherwise you're good to go.
- An approvable letter - quite well done!
You're nearly there and your device seems to pass muster, but the FDA needs more info from you or for you to agree to some conditions. You can amend your application, withdraw it, or, if you really don't agree with this judgment, request an administrative review.
- A non-approvable letter - work to do!
Similar to the approvable letter, but with major amendments required rather than minor tweaks. Your options are the same as those above.
- Denial - oh no!
Flat-out rejection could be caused by non-compliant labeling, false declarations, failure to address past instructions, failure to let the FDA perform extra inspections, or a lapse of compliance in your clinical or non-clinical investigations.
What is the difference between a 510(k) and a PMA?
Because the FDA PMA approval process is designed for high-risk Class III devices, it's more rigorous and longer-lasting than a medium-risk 510(k) submission.
The FDA has 180 days to review your PMA application, double the target length for wrapping up a 510(k) review.
And unfortunately, the average cost of bringing a Class III device to market is around triple that of a Class II device as well. The PMA application fee itself is much steeper, and the adjoining background work of designing and testing your device, sending it through clinical trials, compiling technical data and so on will amount to much more than the time and work required for a Class II marketization.
The 510(k) focuses on proving substantial equivalence between your Class II device and another already on the market: "that one's approved, and we're like that, so we should be approved too".
In contrast, the FDA PMA approval isn't about comparison - it's all about eagle-eyed focus on your device alone. Technical and clinical data forms a much larger part of the PMA submission process, and your device needs to stand on its own two feet as a viable, risk-controlled and efficacious solution.
In general, you'll find PMA approval much harder to achieve than 510(k) clearance.
FURTHER READING: When to submit a 510(k) vs. a PMA
FDA PMA database
Not sure if your device is Class III? Want to get a better feel for the process?
The FDA PMA database is a good starting point as you prepare for your PMA approval prep work.
Visit it to check out approved devices, connected data, attached documents from the submission process, and more.
